
Abstract
Mimetics of the anorexigenic gut hormone glucagon-like peptide 1 (GLP-1) were originally developed as insulinotropic anti-diabetic drugs but also evoke significant weight loss, leading to their recent approval as obesity therapeutics. Co-activation of receptors for GLP-1 and other gut hormones which reduce food intake – peptide YY (PYY3–36), cholecystokinin (CCK) and glucose-dependent insulinotropic peptide (GIP) – is now being explored clinically to enhance efficacy. An alternative approach involves pharmacologically stimulating endogenous secretion of these hormones from enteroendocrine cells (EECs) to recapitulate the metabolic consequences of bariatric surgery, where highly elevated postprandial levels of GLP-1 and PYY3–36 are thought to contribute to improved glycaemia and weight loss.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Obesity is a growing global health concern, and the major modifiable risk factor for type 2 diabetes mellitus (T2DM) and cardiovascular disease. Even relatively modest weight loss (5–10%) can substantially improve insulin sensitivity, pancreatic β-cell function, inflammation, and cardiovascular risk scores (Magkos et al. 2016). Beyond lifestyle modification and poorly-tolerated drugs, the most effective weight loss strategy for common obesity has been bariatric surgery.
Several pharmacological agents targeting the gut hormone axis have recently been developed as novel obesity therapeutics, after successful use in the treatment of T2DM. Once-weekly injections of the long-acting glucagon-like peptide 1 (GLP-1) mimetic semaglutide were recently shown to promote substantial weight loss in obese subjects in a series of phase III trials, with reductions of over 15% observed in half of participants (Wilding et al. 2021). This heralds a new era for obesity therapeutics and encourages development of other medications which activate the same anorexigenic pathways, while minimising side effects. This chapter examines existing and potential approaches to target the enteroendocrine system in obesity, including drugs which directly activate receptors for GLP-1 and other gut hormones, bariatric surgery, and modulation of endogenous enteroendocrine cell (EEC) secretion.
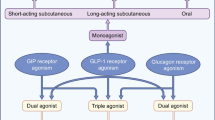
2 Gut Hormone Mimetics: GLP1R Agonism and Beyond
2.1 GLP-1 Receptor Agonists
GLP-1 itself cannot be given therapeutically due to its extremely short half-life (1–2 min), so several injectable GLP1R agonist peptides which are partly resistant to dipeptidyl peptidase 4 (DPP4) degradation have been developed (Drucker and Nauck 2006). Since the approval of the first such GLP1R agonist exenatide in 2005, significant advances have been made to increase half-life, allowing a shift from twice-daily to once-weekly injections and oral formulations (Marso et al. 2016; Zinman et al. 2019). Prominent side effects of incretin mimetics include nausea and GI symptoms such as diarrhoea, although these are somewhat reduced in second-generation treatments and are normally manageable following dose titration (Drucker and Nauck 2006; Zinman et al. 2019).
Following phase III trials showing outstanding weight loss efficacy of 10–20% in overweight subjects, the long-acting GLP1R agonist semaglutide has recently received FDA approval for treatment of obesity (Ryan 2021). This weight loss was far greater than the 5–10% achieved with another GLP1R agonist liraglutide, which was FDA-approved for obesity in 2014 (Mehta et al. 2017). This success is likely to encourage development of next-generation anti-obesity drugs targeting the GLP-1 system, which have the dual benefit of weight loss and improved glycaemic control. GLP1R agonists also reduce the risk of cardiovascular events such as stroke and myocardial infarction (Kristensen et al. 2019), although the underlying mechanisms and the extent of GLP1R expression in the heart and blood vessels remain unclear (McLean et al. 2021).
2.2 DPP4 Inhibitors
Another class of antihyperglycaemic drugs targeting the incretin system are the orally-bioavailable DPP4 inhibitors which increase the circulating half-life of both GLP-1 and the related hormone glucose-dependent insulinotropic peptide (GIP). While DPP4 inhibitors such as sitagliptin increase glucose-stimulated insulin secretion, they do not induce significant weight loss, unlike GLP1R agonists (Drucker and Nauck 2006). DPP4 has a broad range of other substrates, including peptide YY (PYY) and neuropeptide Y, which may have implications for the overall metabolic effects of DPP4 inhibition (Mulvihill and Drucker 2014). DPP4 cleavage converts PYY1–36 to PYY3–36, which mediates the majority of PYY’s anorexigenic effects via NPY2R (Ballantyne 2006). Indeed, the appetite-suppressive effects of PYY1–36 are lost in rats genetically deficient in DPP4 (Unniappan et al. 2006) and activation of NPY1R is linked to increased feeding (Kanatani et al. 2000). It has therefore been proposed that reduced PYY3–36 and increased PYY1–36 levels following DPP4 inhibition account for the lack of weight loss (Aaboe et al. 2010). An alternative possibility, however, would be that intestinal-derived active GLP-1 levels reached under normal physiological conditions are insufficient to promote weight loss. This is supported by findings that the DPP4 inhibitor linagliptin did not enhance anorexia in diet-induced obese (DIO) mice even in the presence of an NPY2R agonist, despite elevated active GLP-1 levels (Hansen et al. 2021), and that GLP1R inhibition did not affect food intake reduction seen following selective chemogenetic activation of distal colonic L-cells, whereas NPY2R inhibition did (Lewis et al. 2020).
2.3 GLP1R/GCGR Co-agonism
In the development of novel incretin mimetics, there is increasing interest in the use of dual or triple agonists targeting other receptors in addition to the GLP1R. The rationale is that these may combine beneficial effects on body weight, glycaemic control and insulin sensitivity while minimising side effects (Brandt et al. 2018). The co-activation of gut hormone receptors also more closely mimics the physiological response to a meal, where multiple enteroendocrine peptides are co-secreted.
Despite the glucose-elevating effects of glucagon (GCG), chronic administration increases energy expenditure, likely via central mechanisms and hepatic upregulation of fibroblast growth factor 21 (FGF21) (Habegger et al. 2013). Glucagon action in the liver also reduces hepatic lipid content and alters hepatocyte metabolism to reduce the extent of non-alcoholic steatohepatitis (NASH), a common obesity co-morbidity (Boland et al. 2020). Balanced synthetic co-agonists of the GLP1R/GCGR, which mimic the activity of oxyntomodulin, are capable of improving glycaemic control in mice and humans (Ambery et al. 2018; Henderson et al. 2016). The GLP1R/GCGR co-agonist SAR425899 induced similar weight loss to liraglutide in a phase IIb trial, while improving HbA1c and increasing β-cell responsiveness to a mixed meal tolerance test (Schiavon et al. 2021); however, this drug was discontinued in 2018 due to incidence of adverse events and disappointing efficacy (Sanofi 2018). A separate phase IIb trial in overweight T2DM patients showed the GLP1R/GCGR co-agonist cotadutide (MEDI0382) evokes slightly greater weight loss (~5%) than a higher dose of liraglutide (Nahra et al. 2021). Unlike liraglutide, cotadutide reduces hepatic fibrosis and lipid content in preclinical NASH models (Boland et al. 2020) and improves markers of hepatic function in T2DM patients (Nahra et al. 2021). The compound BI456906 has also recently entered phase II trials with a direct comparison to semaglutide (Boehringer Ingelheim 2019). Weight loss efficacy of existing GLP1R/GCGR co-agonists does not appear greater than that achieved by best-in-class GLP1R agonists, but these co-agonists may prove particularly useful in the treatment of individuals with obesity and concomitant liver disease.
2.4 GLP1R/GIPR Co-agonism
GIP was long-ignored as a therapeutic target for obesity because of studies linking loss of GIPR signalling to protection from adiposity, and a demonstrated clinical inefficacy in patients with uncontrolled T2DM (Chia et al. 2009; Nauck et al. 1993). Although native GIP and GIPR agonists have at best moderate effects on food intake or body weight (Coskun et al. 2018; Mroz et al. 2019), dual incretin agonists which activate both the GLP1R and GIPR have recently been demonstrated to have greater efficacy in restoring normoglycaemia and reducing body weight than GLP1R agonists alone. The first unimolecular GLP1R/GIPR agonist was the pegylated DPP4-resistant compound NNC0090–2746 (Finan et al. 2013), which evoked similar improvements in HbA1c and body weight to liraglutide in T2DM patients, while also reducing total cholesterol levels (Frias et al. 2017). More striking results have recently been achieved with the dual agonist tirzepatide, a once-weekly injectable peptide biased towards GIPR activation (Coskun et al. 2018). Tirzepatide reduces hyperglycaemia and body weight to a greater extent than the GLP1R agonist comparator dulaglutide, with over a third of T2DM patients achieving over 10% reduction in body weight when treated with the highest dose of tirzepatide (Frias et al. 2018). On-going phase III trials are assessing the cardiovascular outcomes of tirzepatide in T2DM (SURPASS), and the weight loss effects in non-diabetic individuals with obesity (SURMOUNT).
Given this clinical efficacy of dual GLP1R/GIPR agonists, recent work has focused on understanding the underlying mechanisms. In terms of glucose tolerance, improved glycaemic control – which could be initially mediated by GLP1R agonism – is known to re-sensitise pancreatic β-cells to the effects of GIP in T2DM patients (Hojberg et al. 2009). As GIPR antagonists have also been demonstrated to induce weight loss in some (Killion et al. 2018), but not all (Mroz et al. 2019; West et al. 2021), preclinical models, chronic activation of the GIPR has been proposed to cause receptor desensitisation which antagonises endogenous GIP activity (Killion et al. 2020); however, there is currently no substantial evidence to support this hypothesis. Chemogenetic activation of hypothalamic GIPR-expressing cells, which only show a small degree of overlap with neuronal GLP1R expression, acutely reduces food intake in mice (Adriaenssens et al. 2019). A recent publication demonstrated c-fos-staining, marking neuronal activation, in hypothalamic neurons in response to a peripherally administered GIPR agonist; this agonist reduced food intake and weight gain in DIO mice, an effect that was abolished when central GIPR expression was conditionally knocked out through a Nestin-Cre approach (Zhang et al. 2021). The importance of synergistic activation of GIPR- and GLP1R-expressing neurons and downstream anorexigenic pathways, and the potential of GIP for the treatment of obesity in humans, remains an area of active study.
2.5 GLP1R/GCGR/GIPR Triple Agonism
Monomeric drugs which act as triple agonists at the GIP, GLP-1 and GCG receptors have also been developed, in an attempt to combine the beneficial effects of GLP1R/GCGR and GLP1R/GIPR co-agonists. These tri-agonists induce greater body weight loss (up to 30% in DIO mice) alongside improved glucose tolerance and a reversal of steatohepatitis (Finan et al. 2015). Given its effects on liver fat and fibrosis, the tri-agonist HM15211 has now entered phase II trials for the treatment of NASH (Hanmi 2020). Many additional compounds have also been developed and are progressing through preclinical and phase I pipelines for treatment of T2DM (Lilly 2021).
2.6 GLP1R/NPY2R Co-agonism
Several studies have demonstrated additive or synergistic effects of GLP-1 receptor agonists with PYY3–36 (De Silva et al. 2011; Neary et al. 2005; Talsania et al. 2005). In humans, acute (150 min) intravenous infusions of native PYY3–36 in conjunction with GLP-1 reduced energy intake (Schmidt et al. 2014). Similarly, subcutaneous infusion of GLP-1, oxyntomodulin and PYY3–36 over 28 days in overweight individuals improved glycaemic control and caused greater weight loss than semaglutide, but it is impossible to delineate the contribution of each peptide in this study (Behary et al. 2019). High doses of native PYY3–36 induce significant nausea, so doses of PYY analogues would have to be carefully titrated, as is currently done for GLP1R agonists (Gantz et al. 2007). Long-acting analogues of PYY3–36 have been developed and reported to reduce food intake and body weight in primate (Rangwala et al. 2019) and rodent (Lear et al. 2020) models of obesity, alone and in conjunction with GLP1R agonists. Unlike GIP and glucagon, no unimolecular GLP1/PYY3–36 co-agonists have been reported but co-administration of the long-acting PYY analogue PYY 1875 with semaglutide is currently being assessed in phase I human studies for obesity (Novo Nordisk 2019).
2.7 CCK1R Agonism
CCK also has potent anorexigenic effects and several CCK1R agonists were developed in the 2000s as satiety agents (Cawston and Miller 2010). While many of these drugs reduced food intake and body weight in preclinical models, they were deemed no more effective than diet alteration in human studies and were therefore discontinued (Jordan et al. 2008). Co-administration of CCK1R and GLP1R agonists (Trevaskis et al. 2015), or unimolecular co-agonists (Hornigold et al. 2018; Irwin et al. 2015), induces impressive (up to 28%) weight loss in rodent models. A new long-acting CCK1R agonist which reduces food intake and body weight in the obese mini-pig model has recently been developed (Christoffersen et al. 2020). This has not yet progressed to clinical trials but represents a promising compound for use in conjunction with GLP1R agonists if the preclinical efficacy is sustained in longer-term human studies.
2.8 Co-administration of GLP-1 and Amylin Receptor Agonists
Amylin is an anorexigenic hormone co-secreted with insulin from pancreatic β-cells, also expressed in the hypothalamus and, likely at lower levels, in EECs (Boyle et al. 2018; Habib et al. 2012; Li et al. 2015). The area postrema of the brainstem is thought to be critical for the satiety effects of amylin, although other brain regions including the hypothalamic arcuate/ventromedial nuclei and ventral tegmental area may influence control of hedonic feeding (Boyle et al. 2018). Several amylin receptors exist, which are formed when the calcitonin receptor complexes with one or more receptor activity modifying proteins (RAMPs) (Hay et al. 2018). The amylin analogue pramlintide evokes small (typically <5%) reductions in body weight in obese subjects (Aronne et al. 2007). It also induces modest improvements in glycaemic control via inhibition of gastric emptying and suppression of glucagon secretion, and has therefore been approved for treatment of both type 1 and type 2 diabetes (Ryan et al. 2005). A newer once-weekly amylin analogue cagrilintide has demonstrated efficacy in early clinical trials for obesity, alone (phase II) (Fletcher et al. 2021) or in combination with semaglutide (phase Ib) (Enebo et al. 2021). Initial results show a promising additive effect of GLP-1 and amylin receptor agonism, but this has yet to be confirmed in large-scale trials.
2.9 Summary of Gut Hormone Mimetic Treatments
Incretin mimetics have been used clinically for over a decade and are effective in improving glucose tolerance for patients with T2DM with a tolerable safety profile. The potent anorexigenic effects of the once-weekly GLP-1R agonist semaglutide and dual GLP1R/GIPR co-agonist tirzepatide in recent phase III trials represent ground-breaking opportunities for the pharmacological treatment of obesity. These drugs can induce clinically meaningful weight loss of 10–20% in overweight and obese patients, with or without diabetes, and are likely also to confer other beneficial effects such as reduction of cardiovascular mortality. Current pharmaceutical development is focused on combining GLP1R agonism with activity at other hormone receptors to further improve these results, ameliorate other complications of obesity such as NASH and minimise adverse effects.
3 Bariatric Surgery
Currently, bariatric surgical rearrangements of the gastrointestinal tract remain the most successful means of inducing weight loss in patients with severe obesity. Several mechanisms have been proposed to underlie these metabolic changes (reviewed in chapter “Bariatric Surgery”) but a key driver is accelerated nutrient delivery to the distal small intestine and subsequent dramatic increase in secretion of gut hormones such as GLP-1, PYY and oxyntomodulin (Holst et al. 2018; Larraufie et al. 2019). Postprandial excursions of GLP-1, PYY and CCK are strongly elevated immediately post-operatively (Laferrere et al. 2007; Peterli et al. 2012), with exaggerated responses maintained for as long as 20 years after surgery (Naslund et al. 1997). Clinical studies using the GLP1R antagonist exendin-9 demonstrate a clear role for GLP-1 in mediating the beneficial effects of bariatric surgery on postprandial insulin secretion and glucose tolerance (Jorgensen et al. 2013; Larraufie et al. 2019; Salehi et al. 2014), but the importance of post-surgical PYY elevation has been studied in less detail. Following Roux-en-Y gastric bypass (RYGB), double Glp1r/Npy2r knockout mice achieve similar weight loss to wild-type animals (Boland et al. 2019); however, in human RYGB patients the combination of the GLP1R-antagonist exendin-9 and DPP-4 inhibition increases food intake, whilst each treatment alone is ineffective (Svane et al. 2016).
It has been hypothesised that increased circulating GLP-1 and PYY may be the result of altered exposure of the gastrointestinal epithelium to nutrients after surgery leading to changes in stimulus responsiveness or EEC differentiation. Although immunohistochemistry studies in rats (Mumphrey et al. 2013) and humans (Rhee et al. 2015) have reported small changes in L-cell number or density following RYGB, these are insufficient to explain the substantial increase in circulating levels of GLP-1 and PYY. Furthermore, in lean human and mouse models of bariatric surgery there were no major differences in EEC peptide content or transcriptome before and after gastrectomy in humans or mice (Larraufie et al. 2019), suggesting that altered nutrient flow is the critical factor underlying enhanced gut hormone secretion.
4 Stimulating Endogenous EEC Secretion
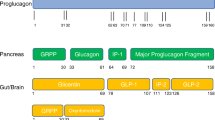
Pharmacologically stimulating endogenous secretion of anorexigenic and insulinotropic hormones or selectively expanding L-cell number could potentially mimic the positive effects of bariatric surgery. In addition to nutrient stimulation, it is possible to directly target the sensory machinery employed by electrically active enteroendocrine cells (Fig. 1). Although existing trials have focused on treating T2DM, the profound weight loss induced by RYGB and GLP-1 receptor agonism is proof of principle that the gut hormone axis is also a promising target for anti-obesity drugs.

Mechanisms of stimulating endogenous enteroendocrine cell secretion
4.1 Nutrient Encapsulation
Initial attempts to stimulate endogenous gut hormone secretion focused on oral delivery of nutritional stimuli. Oral ingestion of the amino acid glutamine modestly increases circulating GLP-1 concentrations (Greenfield et al. 2009) and improves postprandial glycaemic control in T2DM (Samocha-Bonet et al. 2011). Use of a slow-release enteric coating to reduce dosage by selectively delivering glutamine to the L-cell-rich distal gut evoked a small increase in circulating GLP-1 and insulin in fasted individuals, but no metabolic improvements were seen following an oral glucose tolerance test (Meek et al. 2016).
Similar enteric capsules have been used for ileocolonic delivery of free fatty acids and bile acids, in attempt to mimic the rerouting of nutrients observed with bariatric surgery. In fasted T2DM subjects, addition of encapsulated lauric acid to a meal lowers postprandial glucose and evokes a small increase in GLP-1 (Ma et al. 2013). Hydrolysed pine nut oil capsules slightly increase GLP-1 and GIP responses to oral glucose in healthy and overweight volunteers, compared to placebo capsules (Sorensen et al. 2021). Neither study demonstrated notable alterations in circulating insulin levels, likely reflecting inadequate stimulation of incretin release. Acute infusion of the bile salt taurocholate into the rectum potently increases GLP-1, PYY and insulin concentrations, while reducing ad libitum food intake and plasma glucose (Adrian et al. 2012). However, the high doses required for beneficial metabolic effects caused adverse reactions, including rectal irritation and abdominal pain. In a 28-day trial of participants with obesity or T2DM, ileocolonic delivery of conjugated bile acid capsules evoked small but significant improvements in glucose homeostasis, likely mediated by modest increases in GLP-1 (Calderon et al. 2020).
In developing treatments for obesity, it is important to avoid therapeutics with an inherently high caloric load, which would likely negate any beneficial anorexigenic effects. Given the limited success of using nutritional stimuli to evoke secretion of GLP-1, compared to the major elevations following bariatric surgery, efforts have largely shifted towards the use of pharmacological agonists to selectively target receptors on EECs.
4.2 Pharmacological Targeting of Enteroendocrine GPCRs
G-protein coupled receptors (GPCRs) make a significant contribution to regulation of gut hormone secretion in response to products of fat or protein digestion, neurotransmitters and other paracrine or endocrine signals. Activation of EEC receptors coupled to Gq or Gs, leading to Ca2+ and cyclic adenosine monophosphate (cAMP) elevation respectively, is widely recognised to stimulate hormone release. Bile acids strongly evoke gut hormone secretion via the Gs-coupled G-protein bile acid receptor GPBAR1 (Brighton et al. 2015), and monoacylglycerols also act via cAMP signalling downstream of GPR119 (Moss et al. 2016). Several Gq-coupled receptors are important for the postprandial stimulation of gut hormone secretion: the long-chain fatty acid receptors FFA1/GPR40 (Edfalk et al. 2008) and FFA4/GPR120 (Hirasawa et al. 2005), the short chain fatty acid receptor FFA2/GPR43 (Tolhurst et al. 2012), and the aromatic amino acid-responsive calcium sensing receptor CaSR (Pais et al. 2016) and GPR142 (Lin et al. 2016).
Given the inherently druggable nature of GPCRs alongside their cell surface accessibility and relatively localised expression in specific cell populations, these receptors represent a key target in the development of novel therapeutics to enhance endogenous enteroendocrine secretion. Several synthetic compounds which target EEC receptors have been developed to stimulate gut hormone release in attempts to mimic the weight loss effects of bariatric surgery.
4.3 FFA1 (GPR40) Agonists
Agonists of the free fatty acid receptor FFA1/GPR40 were initially developed for their direct effect on pancreatic β-cells (Itoh and Hinuma 2005). The partial agonist TAK-875 showed promising results in T2DM patients, increasing insulin secretion and improving glycaemic control without inducing hypoglycaemia (Burant et al. 2012); however, signs of hepatotoxicity during large-scale phase III trials halted development of TAK-875 (Kaku et al. 2016). There is currently no evidence to suggest these adverse effects were FFA1-mediated (Otieno et al. 2018) and, as liver expression of this receptor is negligible (Briscoe et al. 2003), it is hoped that newer drugs will avoid similar hepatic injury.
An example of a full FFA1 agonist is AM-1638, which evokes GIP and GLP-1 secretion, and causes GLP1R-dependent reductions in plasma glucose in mice (Luo et al. 2012). The structurally-related AM-6226 improves glucose tolerance in a primate model, more effectively than the DPP4 inhibitor sitagliptin (Brown et al. 2018). Other FFA1 agonists in preclinical development which stimulate GLP-1 secretion include SCO-267 (Ueno et al. 2019) and ZYDG2 (Jain et al. 2018). Given its dual role in insulin and incretin secretion, FFA1 presents a promising target for the treatment of T2DM. Whether newer agonists are able to stimulate sufficient release of GLP-1 and other anorexigenic hormones to evoke meaningful effects on body weight remains to be determined.
4.4 GPR119 Agonists
GPR119, the Gs-coupled monoacylglycerol receptor, is also expressed in both enteroendocrine and pancreatic α and β-cells (Chu et al. 2007; Moss et al. 2016). Preclinical studies with the agonist AR231453 demonstrate GPR119-dependent stimulation of GLP-1, GIP and PYY release, and improvements in glucose tolerance which were reduced in Glp1r knockout mice (Chu et al. 2008; Flock et al. 2011). Intriguingly, GPR119 agonism also slows gastric emptying independently of GLP1R, GIPR, GLP-2R and NPY2R (Flock et al. 2011). Nasogastric delivery of the endogenous GPR119 agonist 2-oleoylglycerol in humans induces a small increase in GLP-1 and GIP levels, but this was insufficient to alter insulin or glucose profiles (Hansen et al. 2011).
Despite promising early animal data, the effects of GPR119 agonists on glycaemic control in diabetic populations have been inconclusive. The agonist JNJ-38431055 stimulated GLP-1 and GIP release without altering insulin (Katz et al. 2012). Another agonist GSK1292263 increased circulating PYY approximately five-fold in T2DM subjects but did not affect GLP-1, GIP or glucose levels (Nunez et al. 2014). While this increase in anorexigenic PYY is comparable to postprandial levels after bariatric surgery, there were no differences in self-reported hunger scores. In a phase II trial of Japanese T2DM patients, the agonist DS-8500a improved Hba1c levels, to a lesser extent than sitagliptin, alongside beneficial improvements in lipid and cholesterol (Yamada et al. 2018). The same drug did not alter glycaemic control in a North American cohort when co-dosed with metformin (Sankyo 2018). There are no on-going listed clinical trials of GPR119 agonists for T2DM or obesity, but the agonist MBX-2982 recently entered phase II trials to evaluate whether it can enhance glucagon secretion during insulin-induced hypoglycaemia in type 1 diabetes (CymaBay 2020).
4.5 GPBAR1 (TGR5) Agonists
Activation of the Gs-coupled G-protein bile acid receptor GPBAR1 (previously known as TGR5/GPR131) evokes release of GLP-1, PYY, GIP, GLP-2 and insulin (Bala et al. 2014; Kuhre et al. 2018; Parker et al. 2012b; Thomas et al. 2009). Furthermore, there is preclinical evidence that GPBAR1 agonism may enhance resting brown adipose thermogenesis and decrease inflammation, providing additional benefits in the treatment of obesity (van Nierop et al. 2017).
Bile acids and selective GPBAR1 agonists stimulate GLP-1 secretion and improve glucose homeostasis in wild-type, but not Gpbar1 knockout, mice (Thomas et al. 2009). Colorectal infusion of bile acids also increases PYY/GLP-1 secretion in humans and suppresses appetite in both T2DM and healthy-weight volunteers (Adrian et al. 2012); however, in contrast to animal studies, oral or intrajejunal dosing of bile acids in humans induces very little GLP-1 release with no effect on insulin levels (Hansen et al. 2016; Meyer-Gerspach et al. 2013). In the only published clinical study using a selective GPBAR1 agonist, T2DM subjects were treated with SB-756050 for 6 days (Hodge et al. 2013). This trial produced disappointing effects on glucose tolerance, although there was some indication of increased GLP-1 secretion at certain doses in response to acute GPBAR1 stimulation.
GPBAR1 is widely expressed, and care must therefore be taken to balance the potential therapeutic benefits of receptor activation (improved glucose homeostasis, reduced inflammation) with the risk of diverse off-target effects. These adverse reactions have primarily been studied in animal or cell models but include gallstone formation, cardiovascular alterations, constipation, pruritus and promotion of cell proliferation [reviewed in van Nierop et al. (2017)]. It was initially hoped that it may be possible to target the enteroendocrine cell GPBAR1 directly from the gut lumen with a non-absorbable agonist, to minimise effects in other organ systems; however, it has since been demonstrated that activation of GPBAR1 occurs at the basolateral surface of L-cells, following absorption across the epithelial layer by the apical sodium-dependent bile acid transporter (ASBT) (Brighton et al. 2015).
4.6 Bile Acid Sequestrants and ASBT Inhibitors
Given the challenge of selectively targeting GPBAR1 in EECs pharmacologically, other approaches which alter the intestinal availability of endogenous bile acids may prove effective in the treatment of obesity. The majority (~95%) of bile acids are actively reabsorbed by the ASBT in the distal small intestine and returned to the liver via the portal vein for re-secretion, although a small proportion enter the systemic circulation (Dawson et al. 2009). Bile acids which reach the colon can be modified by the microbiota to form passively-absorbed unconjugated bile acids (Mekhjian et al. 1979), or lost in faeces.
Bile acid sequestrants are non-absorbable resins designed to increase faecal loss of bile acids and therefore increase de novo synthesis to reduce total plasma cholesterol. As well as improving dyslipidaemia, the sequestrant colesevelam also significantly lowers glycaemia in T2DM patients (Bays 2011), an effect which was GPBAR1-dependent in mice (Potthoff et al. 2013). Sequestrants increase delivery of bile acids to the distal intestine and slightly increase postprandial GLP-1 and GIP levels in T2DM patients (Beysen et al. 2012; Brufau et al. 2010); although this is not to the extent seen following bariatric surgery, likely reflecting the need for bile acid absorption for incretin secretion. A recent study of post-RYGB subjects also demonstrated that addition of colesevelam to a meal does not alter circulating GLP-1 levels, arguing against an important role for endogenous bile acids in mediating postprandial GLP-1 release in this cohort (Jonsson et al. 2021).
ASBT inhibitors which block small intestinal bile acid absorption have also been trialled. Although ileal GPBAR1-mediated hormone secretion depends on bile acid absorption via ASBT (Brighton et al. 2015), passive absorption of secondary bile acids in the colon – where there is a large pool of GLP-1 and PYY positive EECs – is likely to still enable GPBAR1 activation (Billing et al. 2019). In diabetic rats, oral administration of ASBT inhibitors promoted GLP-1 and insulin release (Chen et al. 2012). Patients with chronic constipation treated with high doses of the ASBT inhibitor elobixibat for 14 days showed slightly elevated postprandial GLP-1 levels (Rudling et al. 2015). The inhibitor GSK2330672 reduced fasting plasma glucose levels in T2DM subjects, although GLP-1 levels were not measured (Nunez et al. 2016). Notwithstanding gastrointestinal side effects, primarily diarrhoea, ASBT inhibitors are still under development for the treatment of hypercholesterolaemia, NASH and functional constipation. The impact of these drugs on energy intake and body weight should be monitored as they progress through clinical trials, although preclinical studies showed little alteration (Rao et al. 2016).
4.7 Slowing Macronutrient Digestion
Macronutrient breakdown is required for both absorption and stimulation of EEC secretion. Therapies which slow nutrient digestion may therefore reduce the total calories absorbed, while increasing nutrient availability in the GLP-1 and PYY-rich regions of the ileum and colon. Orlistat, a lipase inhibitor approved for the treatment of obesity, reduces breakdown of lipids to fatty acids and monoacylglycerols and delivers a large fat load distally. In healthy volunteers, orlistat taken before a meal reduces circulating GLP-1, PYY and CCK while also reducing satiety (Ellrichmann et al. 2008). The requirement for lipid digestion and subsequent absorption of fatty acids to access basolaterally-located FFA1 (Christensen et al. 2015) appears to limit the ability of orlistat to enhance gut hormone release. Alpha-glucosidase inhibitors, such as acarbose and miglitol, are approved therapies for T2DM which slow the digestion of starch and sucrose to reduce postprandial glucose elevations. These drugs appear to evoke small increases in GLP-1 after a meal or sucrose ingestion, while reducing GIP secretion from the proximal gut (Narita et al. 2012; Seifarth et al. 1998). Postprandial effects on plasma GLP-1 have not been observed in every study (Hucking et al. 2005) and, like orlistat, the efficacy of alpha-glucosidase inhibitors is likely limited by the need for carbohydrate digestion to stimulate EECs in the distal gut.
4.8 SGLT1 Inhibitors
Glucose, from ingested carbohydrates or free sugars, is absorbed across the intestinal epithelium by the apical sodium-dependent glucose cotransporter SGLT1. In electrically active EECs, this coupled uptake of glucose and positively charged sodium ions results in membrane depolarisation and subsequent hormone release (Parker et al. 2012a). Paradoxically, blocking SGLT1 leads to a pronounced increase in circulating GLP-1 at later time points, as well as reducing postprandial glucose excursions (Powell et al. 2017). Following preclinical success, the non-absorbable SGLT1 inhibitor LX-2761 has entered phase I trials as an oral anti-diabetic agent (Lexicon 2018). In parallel to ASBT inhibition, it is hypothesised that blocking proximal uptake increases the delivery of glucose to the distal intestine. As SGLT1 can no longer mediate GLP-1 release in response to glucose, alternative glucose-sensing mechanisms or increased availability of glucose-derived metabolites may be important for EEC activation in this setting.
4.9 Somatostatin Receptor Antagonists
An alternative strategy involves using somatostatin receptor (SSTR) antagonists to release tonic inhibition of enteroendocrine secretion (Jepsen et al. 2019). Specifically, a selective antagonist of SSTR5 – which is highly enriched in ileal L-cells (Moss et al. 2012) – has been shown to improve glycaemic control in mice, in a GLP-1-dependent manner (Jepsen et al. 2021; Sprecher et al. 2010). In rodents, SSTR5 antagonists only improve glucose tolerance when administered orally, further implicating an incretin-like effect rather than direct stimulation of pancreatic insulin secretion (Jepsen et al. 2021). SSTR5 expression does appear to be higher in human than rodent β-cells, and so antagonism may have a dual benefit of stimulating gut hormone and insulin release in man (Farb et al. 2017). Several SSTR antagonists have recently been developed for oral administration (Hirose et al. 2017), but clinical studies for these drugs in obesity or T2DM have not yet been reported.
4.10 Future Directions in Manipulating Endogenous EEC Secretion
The success of L-cell secretagogues in preclinical and clinical studies has been limited to relatively small increases in circulating gut hormone levels. Injectable GLP-1 analogues and bariatric surgery both induce ~ten-fold elevations in concentrations of circulating GLP-1 or equivalents, leading to potent anorexigenic effects (Calara et al. 2005; Yousseif et al. 2014). It appears clear that increasing endogenous gut hormone secretion to these levels will require synergistic activation of multiple pathways. For example, single oral administration of an SSTR5 antagonist, GPBAR1 agonist or FFA1 agonist in mice induces modest (<five-fold) stimulation of GLP-1 secretion (Briere et al. 2018). By contrast, co-treatment with a GPBAR1 or FFA1 agonist (to directly evoke GLP-1 release), a SSTR5 antagonist (to release L-cell inhibition) and a DPP4 inhibitor (to prevent breakdown) elevated circulating GLP-1 to supraphysiological levels, far greater than those achieved post-surgery or during treatment with exogenous incretin mimetics. Similarly, the use of a lipid nanocarrier system for oral exenatide delivery has recently been shown to induce enhanced GLP-1 secretion, synergistically improving glucose tolerance compared to subcutaneous exenatide (Xu et al. 2020). An alternative approach could combine basolateral activation of GPBAR1 or FFA1 from the circulation with a non-caloric drug targeting receptors on the apical processes of open-type EECs, such as the electrogenic glucose transporter SGLT1.
In recent years, bulk and single cell RNA sequencing of human EECs has been carried out using immunolabelled dissociated tissue, or genetically modified reporter organoids (Beumer et al. 2020; Goldspink et al. 2020; Roberts et al. 2019). L-cells express several receptors not previously implicated in postprandial nutrient sensing or neurohormonal regulation, which may represent novel targets for the selective manipulation of endogenous gut hormone secretion.
5 Enhancing EEC Number
Intestinal epithelial cells – exposed to the extreme chemical and physical conditions of the intestinal lumen – have a rapid turnover time, typically only 3–5 days (Darwich et al. 2014). If it were possible to selectively enhance EEC differentiation, this could represent a promising strategy to increase endogenous gut hormone release. Several studies have characterised which transcription factors (TFs) are expressed by specific enteroendocrine cell subtypes and the time course of this expression (Beumer et al. 2020; Gehart et al. 2019). The hormonal repertoire of individual EECs is much broader than suggested by the classical single letter (e.g., ‘L’-cell) classification system and expression of specific hormones is dependent on a combination of factors, including the cell’s location within the gastrointestinal tract, position along the crypt-villus axis and possibly extrinsic influences such as nutritional status. A number of compounds – such as short chain fatty acids and bile acids – have been proposed to physiologically modulate EEC differentiation in organoid and mouse models (Lund et al. 2020; Petersen et al. 2014). Although attempts have been made to develop drugs which boost EEC number (Beumer et al. 2018; Petersen et al. 2015, 2018; Tsakmaki et al. 2020), significant further work is required to selectively target TFs active in specific EEC populations before this approach could be considered for therapeutically increasing gut hormone secretion.
6 Conclusion
As evidenced by recent trials of the GLP-1 receptor agonist semaglutide and the GLP-1/GIP receptor dual agonist tirzepatide, the anorexigenic and insulinotropic gut hormone axes can be effectively manipulated to induce significant weight loss in human subjects. Directly targeting the gut for the treatment of obesity also remains an attractive therapeutic strategy. Pharmacological agonists activating multiple gut hormone receptors, which have demonstrated early clinical success, could be replaced by effective stimulation of plurihormonal EECs. Increasing hormone release from the gut would also enable the activation of local receptors, such as those on vagal afferent neurons, which appear to underlie at least some of the actions of EEC products. By synergistically manipulating several pathways controlling endogenous enteroendocrine secretion, it is theoretically possible to mimic the effectiveness of bariatric surgery in a pill.
References
Aaboe K, Knop FK, Vilsboll T, Deacon CF, Holst JJ, Madsbad S, Krarup T (2010) Twelve weeks treatment with the DPP-4 inhibitor, sitagliptin, prevents degradation of peptide YY and improves glucose and non-glucose induced insulin secretion in patients with type 2 diabetes mellitus. Diabetes Obes Metab 12:323–333. https://doi.org/10.1111/j.1463-1326.2009.01167.x
Adriaenssens AE, Biggs EK, Darwish T, Tadross J, Sukthankar T, Girish M, Polex-Wolf J, Lam BY, Zvetkova I, Pan W, Chiarugi D, Yeo GSH, Blouet C, Gribble FM, Reimann F (2019) Glucose-dependent insulinotropic polypeptide receptor-expressing cells in the hypothalamus regulate food intake. Cell Metab 30:987–996.e6. https://doi.org/10.1016/j.cmet.2019.07.013
Adrian TE, Gariballa S, Parekh KA, Thomas SA, Saadi H, Al Kaabi J, Nagelkerke N, Gedulin B, Young AA (2012) Rectal taurocholate increases L cell and insulin secretion, and decreases blood glucose and food intake in obese type 2 diabetic volunteers. Diabetologia 55:2343–2347. https://doi.org/10.1007/s00125-012-2593-2
Ambery PD, Klammt S, Posch MG, Petrone M, Pu W, Rondinone C, Jermutus L, Hirshberg B (2018) MEDI0382, a GLP-1/glucagon receptor dual agonist, meets safety and tolerability endpoints in a single-dose, healthy-subject, randomized, phase 1 study. Br J Clin Pharmacol 84:2325–2335. https://doi.org/10.1111/bcp.13688
Aronne L, Fujioka K, Aroda V, Chen K, Halseth A, Kesty NC, Burns C, Lush CW, Weyer C (2007) Progressive reduction in body weight after treatment with the amylin analog pramlintide in obese subjects: a phase 2, randomized, placebo-controlled, dose-escalation study. J Clin Endocrinol Metab 92:2977–2983. https://doi.org/10.1210/jc.2006-2003
Bala V, Rajagopal S, Kumar DP, Nalli AD, Mahavadi S, Sanyal AJ, Grider JR, Murthy KS (2014) Release of GLP-1 and PYY in response to the activation of G protein-coupled bile acid receptor TGR5 is mediated by Epac/PLC-epsilon pathway and modulated by endogenous H2S. Front Physiol 5:420. https://doi.org/10.3389/fphys.2014.00420
Ballantyne GH (2006) Peptide YY(1-36) and peptide YY(3-36): part I. distribution, release and actions. Obes Surg 16:651–658. https://doi.org/10.1381/096089206776944959
Bays HE (2011) Colesevelam hydrochloride added to background metformin therapy in patients with type 2 diabetes mellitus: a pooled analysis from 3 clinical studies. Endocr Pract 17:933–938. https://doi.org/10.4158/EP11218.OR
Behary P, Tharakan G, Alexiadou K, Johnson N, Wewer Albrechtsen NJ, Kenkre J, Cuenco J, Hope D, Anyiam O, Choudhury S, Alessimii H, Poddar A, Minnion J, Doyle C, Frost G, Le Roux C, Purkayastha S, Moorthy K, Dhillo W, Holst JJ, Ahmed AR, Prevost AT, Bloom SR, Tan TM (2019) Combined GLP-1, oxyntomodulin, and peptide YY improves body weight and glycemia in obesity and prediabetes/type 2 diabetes: a randomized, single-blinded, placebo-controlled study. Diabetes Care 42:1446–1453. https://doi.org/10.2337/dc19-0449
Beumer J, Artegiani B, Post Y, Reimann F, Gribble F, Nguyen TN, Zeng H, Van den Born M, Van Es JH, Clevers H (2018) Enteroendocrine cells switch hormone expression along the crypt-to-villus BMP signalling gradient. Nat Cell Biol 20(8):909–916
Beumer J, Puschhof J, Bauza-Martinez J, Martinez-Silgado A, Elmentaite R, James KR, Ross A, Hendriks D, Artegiani B, Busslinger GA, Ponsioen B, Andersson-Rolf A, Saftien A, Boot C, Kretzschmar K, Geurts MH, Bar-Ephraim YE, Pleguezuelos-Manzano C, Post Y, Begthel H, van der Linden F, Lopez-Iglesias C, van de Wetering WJ, van der Linden R, Peters PJ, Heck AJR, Goedhart J, Snippert H, Zilbauer M, Teichmann SA, Wu W, Clevers H (2020) High-resolution mRNA and secretome atlas of human enteroendocrine cells. Cell 181(6):1291–1306.e19. https://doi.org/10.1016/j.cell.2020.04.036
Beysen C, Murphy EJ, Deines K, Chan M, Tsang E, Glass A, Turner SM, Protasio J, Riiff T, Hellerstein MK (2012) Effect of bile acid sequestrants on glucose metabolism, hepatic de novo lipogenesis, and cholesterol and bile acid kinetics in type 2 diabetes: a randomised controlled study. Diabetologia 55:432–442. https://doi.org/10.1007/s00125-011-2382-3
Billing LJ, Larraufie P, Lewis J, Leiter A, Li J, Lam B, Yeo GS, Goldspink DA, Kay RG, Gribble FM, Reimann F (2019) Single cell transcriptomic profiling of large intestinal enteroendocrine cells in mice – identification of selective stimuli for insulin-like peptide-5 and glucagon-like peptide-1 co-expressing cells. Mol Metab 29:158–169. https://doi.org/10.1016/j.molmet.2019.09.001
Boehringer Ingelheim (2019) Boehringer Ingelheim and Zealand pharma advance dual-acting GLP-1/glucagon agonist BI 456906 to phase 2 clinical testing in obesity/diabetes
Boland BB, Mumphrey MB, Hao Z, Townsend RL, Gill B, Oldham S, Will S, Morrison CD, Yu S, Munzberg H, Rhodes CJ, Trevaskis JL, Berthoud HR (2019) Combined loss of GLP-1R and Y2R does not alter progression of high-fat diet-induced obesity or response to RYGB surgery in mice. Mol Metab 25:64–72. https://doi.org/10.1016/j.molmet.2019.05.004
Boland ML, Laker RC, Mather K, Nawrocki A, Oldham S, Boland BB, Lewis H, Conway J, Naylor J, Guionaud S, Feigh M, Veidal SS, Lantier L, McGuinness OP, Grimsby J, Rondinone CM, Jermutus L, Larsen MR, Trevaskis JL, Rhodes CJ (2020) Resolution of NASH and hepatic fibrosis by the GLP-1R/GcgR dual-agonist Cotadutide via modulating mitochondrial function and lipogenesis. Nat Metab 2:413–431. https://doi.org/10.1038/s42255-020-0209-6
Boyle CN, Lutz TA, Le Foll C (2018) Amylin – its role in the homeostatic and hedonic control of eating and recent developments of amylin analogs to treat obesity. Mol Metab 8:203–210. https://doi.org/10.1016/j.molmet.2017.11.009
Brandt SJ, Gotz A, Tschop MH, Muller TD (2018) Gut hormone polyagonists for the treatment of type 2 diabetes. Peptides 100:190–201. https://doi.org/10.1016/j.peptides.2017.12.021
Briere DA, Bueno AB, Gunn EJ, Michael MD, Sloop KW (2018) Mechanisms to elevate endogenous GLP-1 beyond injectable GLP-1 analogs and metabolic surgery. Diabetes 67:309–320. https://doi.org/10.2337/db17-0607
Brighton CA, Rievaj J, Kuhre RE, Glass LL, Schoonjans K, Holst JJ, Gribble FM, Reimann F (2015) Bile acids trigger GLP-1 release predominantly by accessing basolaterally located G protein–coupled bile acid receptors. Endocrinology 156:3961–3970. https://doi.org/10.1210/en.2015-1321
Briscoe CP, Tadayyon M, Andrews JL, Benson WG, Chambers JK, Eilert MM, Ellis C, Elshourbagy NA, Goetz AS, Minnick DT, Murdock PR, Sauls HR Jr, Shabon U, Spinage LD, Strum JC, Szekeres PG, Tan KB, Way JM, Ignar DM, Wilson S, Muir AI (2003) The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J Biol Chem 278:11303–11311. https://doi.org/10.1074/jbc.M211495200
Brown SP, Dransfield P, Vimolratana M, Zhu L, Luo J, Zhang J, Jiao X, Pattaropong V, Wong S, Zhuang R, Swaminath G, Houze JB, Lin DC (2018) Discovery of AM-6226: a potent and orally bioavailable GPR40 full agonist that displays efficacy in nonhuman primates. ACS Med Chem Lett 9:757–760. https://doi.org/10.1021/acsmedchemlett.8b00213
Brufau G, Stellaard F, Prado K, Bloks VW, Jonkers E, Boverhof R, Kuipers F, Murphy EJ (2010) Improved glycemic control with colesevelam treatment in patients with type 2 diabetes is not directly associated with changes in bile acid metabolism. Hepatology 52:1455–1464. https://doi.org/10.1002/hep.23831
Burant CF, Viswanathan P, Marcinak J, Cao C, Vakilynejad M, Xie B, Leifke E (2012) TAK-875 versus placebo or glimepiride in type 2 diabetes mellitus: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet 379:1403–1411. https://doi.org/10.1016/S0140-6736(11)61879-5
Calara F, Taylor K, Han J, Zabala E, Carr EM, Wintle M, Fineman M (2005) A randomized, open-label, crossover study examining the effect of injection site on bioavailability of exenatide (synthetic exendin-4). Clin Ther 27:210–215. https://doi.org/10.1016/j.clinthera.2005.02.008
Calderon G, McRae A, Rievaj J, Davis J, Zandvakili I, Linker-Nord S, Burton D, Roberts G, Reimann F, Gedulin B, Vella A, LaRusso NF, Camilleri M, Gribble FM, Acosta A (2020) Ileo-colonic delivery of conjugated bile acids improves glucose homeostasis via colonic GLP-1-producing enteroendocrine cells in human obesity and diabetes. EBioMedicine 55:102759. https://doi.org/10.1016/j.ebiom.2020.102759
Cawston EE, Miller LJ (2010) Therapeutic potential for novel drugs targeting the type 1 cholecystokinin receptor. Br J Pharmacol 159:1009–1021. https://doi.org/10.1111/j.1476-5381.2009.00489.x
Chen L, Yao X, Young A, McNulty J, Anderson D, Liu Y, Nystrom C, Croom D, Ross S, Collins J, Rajpal D, Hamlet K, Smith C, Gedulin B (2012) Inhibition of apical sodium-dependent bile acid transporter as a novel treatment for diabetes. Am J Physiol Endocrinol Metab 302:E68–E76. https://doi.org/10.1152/ajpendo.00323.2011
Chia CW, Carlson OD, Kim W, Shin YK, Charles CP, Kim HS, Melvin DL, Egan JM (2009) Exogenous glucose-dependent insulinotropic polypeptide worsens post prandial hyperglycemia in type 2 diabetes. Diabetes 58:1342–1349. https://doi.org/10.2337/db08-0958
Christensen LW, Kuhre RE, Janus C, Svendsen B, Holst JJ (2015) Vascular, but not luminal, activation of FFAR1 (GPR40) stimulates GLP-1 secretion from isolated perfused rat small intestine. Physiol Rep 3:e12551. https://doi.org/10.14814/phy2.12551
Christoffersen BO, Skyggebjerg RB, Bugge A, Kirk RK, Vestergaard B, Uldam HK, Fels JJ, Pyke C, Sensfuss U, Sanfridson A, Clausen TR (2020) Long-acting CCK analogue NN9056 lowers food intake and body weight in obese Gottingen Minipigs. Int J Obes 44:447–456. https://doi.org/10.1038/s41366-019-0386-0
Chu ZL, Jones RM, He H, Carroll C, Gutierrez V, Lucman A, Moloney M, Gao H, Mondala H, Bagnol D, Unett D, Liang Y, Demarest K, Semple G, Behan DP, Leonard J (2007) A role for beta-cell-expressed G protein-coupled receptor 119 in glycemic control by enhancing glucose-dependent insulin release. Endocrinology 148:2601–2609. https://doi.org/10.1210/en.2006-1608
Chu ZL, Carroll C, Alfonso J, Gutierrez V, He H, Lucman A, Pedraza M, Mondala H, Gao H, Bagnol D, Chen R, Jones RM, Behan DP, Leonard J (2008) A role for intestinal endocrine cell-expressed g protein-coupled receptor 119 in glycemic control by enhancing glucagon-like Peptide-1 and glucose-dependent insulinotropic peptide release. Endocrinology 149:2038–2047. https://doi.org/10.1210/en.2007-0966
Coskun T, Sloop KW, Loghin C, Alsina-Fernandez J, Urva S, Bokvist KB, Cui X, Briere DA, Cabrera O, Roell WC, Kuchibhotla U, Moyers JS, Benson CT, Gimeno RE, D'Alessio DA, Haupt A (2018) LY3298176, a novel dual GIP and GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus: from discovery to clinical proof of concept. Mol Metab 18:3–14. https://doi.org/10.1016/j.molmet.2018.09.009
CymaBay (2020) CymaBay announces study to evaluate the potential for GPR119 agonists to prevent hypoglycemia in type 1 diabetes. https://www.globenewswire.com/en/news-release/2020/11/05/2120994/37067/en/CymaBay-Announces-Study-to-Evaluate-the-Potential-for-GPR119-Agonists-to-Prevent-Hypoglycemia-in-Type-1-Diabetes.html. Accessed 1 Jun 2021
Darwich AS, Aslam U, Ashcroft DM, Rostami-Hodjegan A (2014) Meta-analysis of the turnover of intestinal epithelia in preclinical animal species and humans. Drug Metab Dispos 42:2016–2022. https://doi.org/10.1124/dmd.114.058404
Dawson PA, Lan T, Rao A (2009) Bile acid transporters. J Lipid Res 50:2340–2357. https://doi.org/10.1194/jlr.R900012-JLR200
De Silva A, Salem V, Long CJ, Makwana A, Newbould RD, Rabiner EA, Ghatei MA, Bloom SR, Matthews PM, Beaver JD, Dhillo WS (2011) The gut hormones PYY 3-36 and GLP-1 7-36 amide reduce food intake and modulate brain activity in appetite centers in humans. Cell Metab 14:700–706. https://doi.org/10.1016/j.cmet.2011.09.010
Drucker DJ, Nauck MA (2006) The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 368:1696–1705. https://doi.org/10.1016/S0140-6736(06)69705-5
Edfalk S, Steneberg P, Edlund H (2008) Gpr40 is expressed in enteroendocrine cells and mediates free fatty acid stimulation of incretin secretion. Diabetes 57:2280–2287. https://doi.org/10.2337/db08-0307
Ellrichmann M, Kapelle M, Ritter PR, Holst JJ, Herzig KH, Schmidt WE, Schmitz F, Meier JJ (2008) Orlistat inhibition of intestinal lipase acutely increases appetite and attenuates postprandial glucagon-like peptide-1-(7-36)-amide-1, cholecystokinin, and peptide YY concentrations. J Clin Endocrinol Metab 93:3995–3998. https://doi.org/10.1210/jc.2008-0924
Enebo LB, Berthelsen KK, Kankam M, Lund MT, Rubino DM, Satylganova A, Lau DCW (2021) Safety, tolerability, pharmacokinetics, and pharmacodynamics of concomitant administration of multiple doses of cagrilintide with semaglutide 2.4 mg for weight management: a randomised, controlled, phase 1b trial. Lancet 397:1736–1748. https://doi.org/10.1016/S0140-6736(21)00845-X
Farb TB, Adeva M, Beauchamp TJ, Cabrera O, Coates DA, Meredith TD, Droz BA, Efanov A, Ficorilli JV, Gackenheimer SL, Martinez-Grau MA, Molero V, Ruano G, Statnick MA, Suter TM, Syed SK, Toledo MA, Willard FS, Zhou X, Bokvist KB, Barrett DG (2017) Regulation of endogenous (male) rodent GLP-1 secretion and human islet insulin secretion by antagonism of somatostatin receptor 5. Endocrinology 158:3859–3873. https://doi.org/10.1210/en.2017-00639
Finan B, Ma T, Ottaway N, Muller TD, Habegger KM, Heppner KM, Kirchner H, Holland J, Hembree J, Raver C, Lockie SH, Smiley DL, Gelfanov V, Yang B, Hofmann S, Bruemmer D, Drucker DJ, Pfluger PT, Perez-Tilve D, Gidda J, Vignati L, Zhang L, Hauptman JB, Lau M, Brecheisen M, Uhles S, Riboulet W, Hainaut E, Sebokova E, Conde-Knape K, Konkar A, DiMarchi RD, Tschop MH (2013) Unimolecular dual incretins maximize metabolic benefits in rodents, monkeys, and humans. Sci Transl Med 5:209ra151. https://doi.org/10.1126/scitranslmed.3007218
Finan B, Yang B, Ottaway N, Smiley DL, Ma T, Clemmensen C, Chabenne J, Zhang L, Habegger KM, Fischer K, Campbell JE, Sandoval D, Seeley RJ, Bleicher K, Uhles S, Riboulet W, Funk J, Hertel C, Belli S, Sebokova E, Conde-Knape K, Konkar A, Drucker DJ, Gelfanov V, Pfluger PT, Muller TD, Perez-Tilve D, DiMarchi RD, Tschop MH (2015) A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat Med 21:27–36. https://doi.org/10.1038/nm.3761
Fletcher MM, Keov P, Truong TT, Mennen G, Hick CA, Zhao P, Furness SGB, Kruse T, Clausen TR, Wootten D, Sexton PM (2021) AM833 is a novel agonist of calcitonin family G protein-coupled receptors: pharmacological comparison with six selective and nonselective agonists. J Pharmacol Exp Ther 377:417–440. https://doi.org/10.1124/jpet.121.000567
Flock G, Holland D, Seino Y, Drucker DJ (2011) GPR119 regulates murine glucose homeostasis through incretin receptor-dependent and independent mechanisms. Endocrinology 152:374–383. https://doi.org/10.1210/en.2010-1047
Frias JP, Bastyr EJ 3rd, Vignati L, Tschop MH, Schmitt C, Owen K, Christensen RH, DiMarchi RD (2017) The sustained effects of a dual GIP/GLP-1 receptor agonist, NNC0090-2746, in patients with type 2 diabetes. Cell Metab 26:343–352.e2. https://doi.org/10.1016/j.cmet.2017.07.011
Frias JP, Nauck MA, Van J, Kutner ME, Cui X, Benson C, Urva S, Gimeno RE, Milicevic Z, Robins D, Haupt A (2018) Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: a randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet 392:2180–2193. https://doi.org/10.1016/S0140-6736(18)32260-8
Gantz I, Erondu N, Mallick M, Musser B, Krishna R, Tanaka WK, Snyder K, Stevens C, Stroh MA, Zhu H, Wagner JA, Macneil DJ, Heymsfield SB, Amatruda JM (2007) Efficacy and safety of intranasal peptide YY3-36 for weight reduction in obese adults. J Clin Endocrinol Metab 92:1754–1757. https://doi.org/10.1210/jc.2006-1806
Gehart H, van Es JH, Hamer K, Beumer J, Kretzschmar K, Dekkers JF, Rios A, Clevers H (2019) Identification of enteroendocrine regulators by real-time single-cell differentiation mapping. Cell 176:1158–1173.e16. https://doi.org/10.1016/j.cell.2018.12.029
Goldspink DA, Lu VB, Miedzybrodzka EL, Smith CA, Foreman RE, Billing LJ, Kay RG, Reimann F, Gribble FM (2020) Labeling and characterization of human GLP-1-secreting L-cells in primary ileal organoid culture. Cell Rep 31:107833. https://doi.org/10.1016/j.celrep.2020.107833
Greenfield JR, Farooqi IS, Keogh JM, Henning E, Habib AM, Blackwood A, Reimann F, Holst JJ, Gribble FM (2009) Oral glutamine increases circulating glucagon-like peptide 1, glucagon, and insulin concentrations in lean, obese, and type 2 diabetic subjects. Am J Clin Nutr 89:106–113. https://doi.org/10.3945/ajcn.2008.26362
Habegger KM, Stemmer K, Cheng C, Muller TD, Heppner KM, Ottaway N, Holland J, Hembree JL, Smiley D, Gelfanov V, Krishna R, Arafat AM, Konkar A, Belli S, Kapps M, Woods SC, Hofmann SM, D'Alessio D, Pfluger PT, Perez-Tilve D, Seeley RJ, Konishi M, Itoh N, Kharitonenkov A, Spranger J, DiMarchi RD, Tschop MH (2013) Fibroblast growth factor 21 mediates specific glucagon actions. Diabetes 62:1453–1463. https://doi.org/10.2337/db12-1116
Habib AM, Richards P, Cairns LS, Rogers GJ, Bannon CAM, Parker HE, Morley TCE, Yeo GSH, Reimann F, Gribble FM (2012) Overlap of endocrine hormone expression in the mouse intestine revealed by transcriptional profiling and flow cytometry. Endocrinology 153:3054–3065. https://doi.org/10.1210/en.2011-2170
Hanmi (2020) Study to evaluate efficacy, safety and tolerability of HM15211 in subjects. https://clinicaltrials.gov/ct2/show/NCT04505436. Accessed 3 Jun 2021
Hansen KB, Rosenkilde MM, Knop FK, Wellner N, Diep TA, Rehfeld JF, Andersen UB, Holst JJ, Hansen HS (2011) 2-Oleoyl glycerol is a GPR119 agonist and signals GLP-1 release in humans. J Clin Endocrinol Metab 96:E1409–E1417. https://doi.org/10.1210/jc.2011-0647
Hansen M, Scheltema MJ, Sonne DP, Hansen JS, Sperling M, Rehfeld JF, Holst JJ, Vilsboll T, Knop FK (2016) Effect of chenodeoxycholic acid and the bile acid sequestrant colesevelam on glucagon-like peptide-1 secretion. Diabetes Obes Metab 18:571–580. https://doi.org/10.1111/dom.12648
Hansen HH, Gronlund RV, Baader-Pagler T, Haebel P, Tammen H, Larsen LK, Jelsing J, Vrang N, Klein T (2021) Characterization of combined linagliptin and Y2R agonist treatment in diet-induced obese mice. Sci Rep 11:8060. https://doi.org/10.1038/s41598-021-87539-7
Hay DL, Garelja ML, Poyner DR, Walker CS (2018) Update on the pharmacology of calcitonin/CGRP family of peptides: IUPHAR review 25. Br J Pharmacol 175:3–17. https://doi.org/10.1111/bph.14075
Henderson SJ, Konkar A, Hornigold DC, Trevaskis JL, Jackson R, Fritsch Fredin M, Jansson-Lofmark R, Naylor J, Rossi A, Bednarek MA, Bhagroo N, Salari H, Will S, Oldham S, Hansen G, Feigh M, Klein T, Grimsby J, Maguire S, Jermutus L, Rondinone CM, Coghlan MP (2016) Robust anti-obesity and metabolic effects of a dual GLP-1/glucagon receptor peptide agonist in rodents and non-human primates. Diabetes Obes Metab 18:1176–1190. https://doi.org/10.1111/dom.12735
Hirasawa A, Tsumaya K, Awaji T, Katsuma S, Adachi T, Yamada M, Sugimoto Y, Miyazaki S, Tsujimoto G (2005) Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat Med 11:90–94. https://doi.org/10.1038/nm1168
Hirose H, Yamasaki T, Ogino M, Mizojiri R, Tamura-Okano Y, Yashiro H, Muraki Y, Nakano Y, Sugama J, Hata A, Iwasaki S, Watanabe M, Maekawa T, Kasai S (2017) Discovery of novel 5-oxa-2,6-diazaspiro[3.4]oct-6-ene derivatives as potent, selective, and orally available somatostatin receptor subtype 5 (SSTR5) antagonists for treatment of type 2 diabetes mellitus. Bioorg Med Chem 25:4175–4193. https://doi.org/10.1016/j.bmc.2017.06.007
Hodge RJ, Lin J, Vasist Johnson LS, Gould EP, Bowers GD, Nunez DJ, Team SBP (2013) Safety, pharmacokinetics, and pharmacodynamic effects of a selective TGR5 agonist, SB-756050, in type 2 diabetes. Clin Pharmacol Drug Dev 2:213–222. https://doi.org/10.1002/cpdd.34
Hojberg PV, Vilsboll T, Rabol R, Knop FK, Bache M, Krarup T, Holst JJ, Madsbad S (2009) Four weeks of near-normalisation of blood glucose improves the insulin response to glucagon-like peptide-1 and glucose-dependent insulinotropic polypeptide in patients with type 2 diabetes. Diabetologia 52:199–207. https://doi.org/10.1007/s00125-008-1195-5
Holst JJ, Madsbad S, Bojsen-Moller KN, Svane MS, Jorgensen NB, Dirksen C, Martinussen C (2018) Mechanisms in bariatric surgery: gut hormones, diabetes resolution, and weight loss. Surg Obes Relat Dis 14:708–714. https://doi.org/10.1016/j.soard.2018.03.003
Hornigold DC, Roth E, Howard V, Will S, Oldham S, Coghlan MP, Blouet C, Trevaskis JL (2018) A GLP-1:CCK fusion peptide harnesses the synergistic effects on metabolism of CCK-1 and GLP-1 receptor agonism in mice. Appetite 127:334–340. https://doi.org/10.1016/j.appet.2018.05.131
Hucking K, Kostic Z, Pox C, Ritzel R, Holst JJ, Schmiegel W, Nauck MA (2005) Alpha-glucosidase inhibition (acarbose) fails to enhance secretion of glucagon-like peptide 1 (7-36 amide) and to delay gastric emptying in type 2 diabetic patients. Diabet Med 22:470–476. https://doi.org/10.1111/j.1464-5491.2005.01451.x
Irwin N, Pathak V, Flatt PR (2015) A novel CCK-8/GLP-1 hybrid peptide exhibiting prominent insulinotropic, glucose-lowering, and satiety actions with significant therapeutic potential in high-fat-fed mice. Diabetes 64:2996–3009. https://doi.org/10.2337/db15-0220
Itoh Y, Hinuma S (2005) GPR40, a free fatty acid receptor on pancreatic beta cells, regulates insulin secretion. Hepatol Res 33:171–173. https://doi.org/10.1016/j.hepres.2005.09.028
Jain MR, Giri SR, Trivedi CJ, Bhoi BB, Rath AC, Rathod RM, Sundar R, Bandyopadhyay D, Ramdhave R, Patel GD, Srivastava BK, Desai RC (2018) ZYDG2, a potent, selective, and safe GPR40 agonist for treatment of type 2 diabetes. Diabetes 67. https://doi.org/10.2337/db18-1194-P
Jepsen SL, Grunddal KV, Wewer Albrechtsen NJ, Engelstoft MS, Gabe MBN, Jensen EP, Orskov C, Poulsen SS, Rosenkilde MM, Pedersen J, Gribble FM, Reimann F, Deacon CF, Schwartz TW, Christ AD, Martin RE, Holst JJ (2019) Paracrine crosstalk between intestinal L- and D-cells controls secretion of glucagon-like peptide-1 in mice. Am J Physiol Endocrinol Metab 317:E1081–E1093. https://doi.org/10.1152/ajpendo.00239.2019
Jepsen SL, Albrechtsen NJW, Windelov JA, Galsgaard KD, Hunt JE, Farb TB, Kissow H, Pedersen J, Deacon CF, Martin RE, Holst JJ (2021) Antagonizing somatostatin receptor subtype 2 and 5 reduces blood glucose in a gut- and GLP-1R-dependent manner. JCI Insight 6. https://doi.org/10.1172/jci.insight.143228
Jonsson I, Bojsen-Moller KN, Kristiansen VB, Veedfald S, Wewer Albrechtsen NJ, Clausen TR, Kuhre RE, Rehfeld JF, Holst JJ, Madsbad S, Svane MS (2021) Effects of manipulating circulating bile acid concentrations on postprandial GLP-1 secretion and glucose metabolism after Roux-en-Y gastric bypass. Front Endocrinol (Lausanne) 12:681116. https://doi.org/10.3389/fendo.2021.681116
Jordan J, Greenway FL, Leiter LA, Li Z, Jacobson P, Murphy K, Hill J, Kler L, Aftring RP (2008) Stimulation of cholecystokinin-a receptors with GI181771X does not cause weight loss in overweight or obese patients. Clin Pharmacol Ther 83:281–287. https://doi.org/10.1038/sj.clpt.6100272
Jorgensen NB, Dirksen C, Bojsen-Moller KN, Jacobsen SH, Worm D, Hansen DL, Kristiansen VB, Naver L, Madsbad S, Holst JJ (2013) Exaggerated glucagon-like peptide 1 response is important for improved beta-cell function and glucose tolerance after Roux-en-Y gastric bypass in patients with type 2 diabetes. Diabetes 62:3044–3052. https://doi.org/10.2337/db13-0022
Kaku K, Enya K, Nakaya R, Ohira T, Matsuno R (2016) Long-term safety and efficacy of fasiglifam (TAK-875), a G-protein-coupled receptor 40 agonist, as monotherapy and combination therapy in Japanese patients with type 2 diabetes: a 52-week open-label phase III study. Diabetes Obes Metab 18:925–929. https://doi.org/10.1111/dom.12693
Kanatani A, Mashiko S, Murai N, Sugimoto N, Ito J, Fukuroda T, Fukami T, Morin N, MacNeil DJ, Van der Ploeg LH, Saga Y, Nishimura S, Ihara M (2000) Role of the Y1 receptor in the regulation of neuropeptide Y-mediated feeding: comparison of wild-type, Y1 receptor-deficient, and Y5 receptor-deficient mice. Endocrinology 141:1011–1016. https://doi.org/10.1210/endo.141.3.7387
Katz LB, Gambale JJ, Rothenberg PL, Vanapalli SR, Vaccaro N, Xi L, Sarich TC, Stein PP (2012) Effects of JNJ-38431055, a novel GPR119 receptor agonist, in randomized, double-blind, placebo-controlled studies in subjects with type 2 diabetes. Diabetes Obes Metab 14:709–716. https://doi.org/10.1111/j.1463-1326.2012.01587.x
Killion EA, Wang J, Yie J, Shi SD, Bates D, Min X, Komorowski R, Hager T, Deng L, Atangan L, Lu SC, Kurzeja RJM, Sivits G, Lin J, Chen Q, Wang Z, Thibault SA, Abbott CM, Meng T, Clavette B, Murawsky CM, Foltz IN, Rottman JB, Hale C, Veniant MM, Lloyd DJ (2018) Anti-obesity effects of GIPR antagonists alone and in combination with GLP-1R agonists in preclinical models. Sci Transl Med 10. https://doi.org/10.1126/scitranslmed.aat3392
Killion EA, Lu SC, Fort M, Yamada Y, Veniant MM, Lloyd DJ (2020) Glucose-dependent insulinotropic polypeptide receptor therapies for the treatment of obesity, do agonists = antagonists? Endocr Rev 41. https://doi.org/10.1210/endrev/bnz002
Kristensen SL, Rorth R, Jhund PS, Docherty KF, Sattar N, Preiss D, Kober L, Petrie MC, McMurray JJV (2019) Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials. Lancet Diabetes Endocrinol 7:776–785. https://doi.org/10.1016/S2213-8587(19)30249-9
Kuhre RE, Wewer Albrechtsen NJ, Larsen O, Jepsen SL, Balk-Moller E, Andersen DB, Deacon CF, Schoonjans K, Reimann F, Gribble FM, Albrechtsen R, Hartmann B, Rosenkilde MM, Holst JJ (2018) Bile acids are important direct and indirect regulators of the secretion of appetite- and metabolism-regulating hormones from the gut and pancreas. Mol Metab 11:84–95. https://doi.org/10.1016/j.molmet.2018.03.007
Laferrere B, Heshka S, Wang K, Khan Y, McGinty J, Teixeira J, Hart AB, Olivan B (2007) Incretin levels and effect are markedly enhanced 1 month after Roux-en-Y gastric bypass surgery in obese patients with type 2 diabetes. Diabetes Care 30:1709–1716. https://doi.org/10.2337/dc06-1549
Larraufie P, Roberts GP, McGavigan AK, Kay RG, Li J, Leiter A, Melvin A, Biggs EK, Ravn P, Davy K, Hornigold DC, Yeo GSH, Hardwick RH, Reimann F, Gribble FM (2019) Important role of the GLP-1 axis for glucose homeostasis after bariatric surgery. Cell Rep 26:1399–1408.e6. https://doi.org/10.1016/j.celrep.2019.01.047
Lear S, Pflimlin E, Zhou Z, Huang D, Weng S, Nguyen-Tran V, Joseph SB, Roller S, Peterson S, Li J, Tremblay M, Schultz PG, Shen W (2020) Engineering of a potent, long-acting NPY2R agonist for combination with a GLP-1R agonist as a multi-hormonal treatment for obesity. J Med Chem 63:9660–9671. https://doi.org/10.1021/acs.jmedchem.0c00740
Lewis JE, Miedzybrodzka EL, Foreman RE, Woodward ORM, Kay RG, Goldspink DA, Gribble FM, Reimann F (2020) Selective stimulation of colonic L cells improves metabolic outcomes in mice. Diabetologia. https://doi.org/10.1007/s00125-020-05149-w
Lexicon (2018) Lexicon Pharmaceuticals announces topline phase 1 clinical results for LX2761 in diabetes. https://www.lexpharma.com/media-center/news/693-lexicon-pharmaceuticals-announces-topline-phase-1-clinical-results-for-lx2761-in-diabetes. Accessed 4 Jun 2021
Li Z, Kelly L, Heiman M, Greengard P, Friedman JM (2015) Hypothalamic amylin acts in concert with leptin to regulate food intake. Cell Metab 22:1059–1067. https://doi.org/10.1016/j.cmet.2015.10.012
Lilly (2021) Medicines in development. https://www.lilly.com/discovery/clinical-development-pipeline. Accessed 1 Jun 2021
Lin HV, Efanov AM, Fang X, Beavers LS, Wang X, Wang J, Gonzalez Valcarcel IC, Ma T (2016) GPR142 controls tryptophan-induced insulin and incretin hormone secretion to improve glucose metabolism. PLoS One 11:e0157298. https://doi.org/10.1371/journal.pone.0157298
Lund ML, Sorrentino G, Egerod KL, Kroone C, Mortensen B, Knop FK, Reimann F, Gribble FM, Drucker DJ, de Koning EJP, Schoonjans K, Backhed F, Schwartz TW, Petersen N (2020) L-cell differentiation is induced by bile acids through GPBAR1 and paracrine GLP-1 and serotonin signaling. Diabetes 69:614–623. https://doi.org/10.2337/db19-0764
Luo J, Swaminath G, Brown SP, Zhang J, Guo Q, Chen M, Nguyen K, Tran T, Miao L, Dransfield PJ, Vimolratana M, Houze JB, Wong S, Toteva M, Shan B, Li F, Zhuang R, Lin DC (2012) A potent class of GPR40 full agonists engages the enteroinsular axis to promote glucose control in rodents. PLoS One 7:e46300. https://doi.org/10.1371/journal.pone.0046300
Ma J, Checklin HL, Wishart JM, Stevens JE, Jones KL, Horowitz M, Meyer JH, Rayner CK (2013) A randomised trial of enteric-coated nutrient pellets to stimulate gastrointestinal peptide release and lower glycaemia in type 2 diabetes. Diabetologia 56:1236–1242. https://doi.org/10.1007/s00125-013-2876-2
Magkos F, Fraterrigo G, Yoshino J, Luecking C, Kirbach K, Kelly SC, de Las FL, He S, Okunade AL, Patterson BW, Klein S (2016) Effects of moderate and subsequent progressive weight loss on metabolic function and adipose tissue biology in humans with obesity. Cell Metab 23:591–601. https://doi.org/10.1016/j.cmet.2016.02.005
Marso SP, Bain SC, Consoli A, Eliaschewitz FG, Jodar E, Leiter LA, Lingvay I, Rosenstock J, Seufert J, Warren ML, Woo V, Hansen O, Holst AG, Pettersson J, Vilsboll T, SUSTAIN-6 Investigators (2016) Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med 375:1834–1844. https://doi.org/10.1056/NEJMoa1607141
McLean BA, Wong CK, Campbell JE, Hodson DJ, Trapp S, Drucker DJ (2021) Revisiting the complexity of GLP-1 action from sites of synthesis to receptor activation. Endocr Rev 42:101–132. https://doi.org/10.1210/endrev/bnaa032
Meek CL, Lewis HB, Vergese B, Park A, Reimann F, Gribble F (2016) The effect of encapsulated glutamine on gut peptide secretion in human volunteers. Peptides 77:38–46. https://doi.org/10.1016/j.peptides.2015.10.008
Mehta A, Marso SP, Neeland IJ (2017) Liraglutide for weight management: a critical review of the evidence. Obes Sci Pract 3:3–14. https://doi.org/10.1002/osp4.84
Mekhjian HS, Phillips SF, Hofmann AF (1979) Colonic absorption of unconjugated bile acids: perfusion studies in man. Dig Dis Sci 24:545–550. https://doi.org/10.1007/BF01489324
Meyer-Gerspach AC, Steinert RE, Keller S, Malarski A, Schulte FH, Beglinger C (2013) Effects of chenodeoxycholic acid on the secretion of gut peptides and fibroblast growth factors in healthy humans. J Clin Endocrinol Metab 98:3351–3358. https://doi.org/10.1210/jc.2012-4109
Moss CE, Marsh WJ, Parker HE, Ogunnowo-Bada E, Riches CH, Habib AM, Evans ML, Gribble FM, Reimann F (2012) Somatostatin receptor 5 and cannabinoid receptor 1 activation inhibit secretion of glucose-dependent insulinotropic polypeptide from intestinal K cells in rodents. Diabetologia 55:3094–3103. https://doi.org/10.1007/s00125-012-2663-5
Moss CE, Glass LL, Diakogiannaki E, Pais R, Lenaghan C, Smith DM, Wedin M, Bohlooly-Y M, Gribble FM, Reimann F (2016) Lipid derivatives activate GPR119 and trigger GLP-1 secretion in primary murine L-cells. Peptides 77:16–20. https://doi.org/10.1016/J.PEPTIDES.2015.06.012
Mroz PA, Finan B, Gelfanov V, Yang B, Tschop MH, DiMarchi RD, Perez-Tilve D (2019) Optimized GIP analogs promote body weight lowering in mice through GIPR agonism not antagonism. Mol Metab 20:51–62. https://doi.org/10.1016/j.molmet.2018.12.001
Mulvihill EE, Drucker DJ (2014) Pharmacology, physiology, and mechanisms of action of dipeptidyl peptidase-4 inhibitors. Endocr Rev 35:992–1019. https://doi.org/10.1210/er.2014-1035
Mumphrey MB, Patterson LM, Zheng H, Berthoud HR (2013) Roux-en-Y gastric bypass surgery increases number but not density of CCK-, GLP-1-, 5-HT-, and neurotensin-expressing enteroendocrine cells in rats. Neurogastroenterol Motil 25:e70–e79. https://doi.org/10.1111/nmo.12034
Nahra R, Wang T, Gadde KM, Oscarsson J, Stumvoll M, Jermutus L, Hirshberg B, Ambery P (2021) Effects of Cotadutide on metabolic and hepatic parameters in adults with overweight or obesity and type 2 diabetes: a 54-week randomized phase 2b study. Diabetes Care. https://doi.org/10.2337/dc20-2151
Narita T, Yokoyama H, Yamashita R, Sato T, Hosoba M, Morii T, Fujita H, Tsukiyama K, Yamada Y (2012) Comparisons of the effects of 12-week administration of miglitol and voglibose on the responses of plasma incretins after a mixed meal in Japanese type 2 diabetic patients. Diabetes Obes Metab 14:283–287. https://doi.org/10.1111/j.1463-1326.2011.01526.x
Naslund E, Gryback P, Hellstrom PM, Jacobsson H, Holst JJ, Theodorsson E, Backman L (1997) Gastrointestinal hormones and gastric emptying 20 years after jejunoileal bypass for massive obesity. Int J Obes Relat Metab Disord 21:387–392. https://doi.org/10.1038/sj.ijo.0800418
Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W (1993) Preserved incretin activity of glucagon-like peptide 1 [7-36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest 91:301–307. https://doi.org/10.1172/JCI116186
Neary NM, Small CJ, Druce MR, Park AJ, Ellis SM, Semjonous NM, Dakin CL, Filipsson K, Wang F, Kent AS, Frost GS, Ghatei MA, Bloom SR (2005) Peptide YY3-36 and glucagon-like peptide-17-36 inhibit food intake additively. Endocrinology 146:5120–5127. https://doi.org/10.1210/en.2005-0237
Novo Nordisk (2019) Research study of a new medicine (NNC9204-1706) in people with overweight or obesity. https://clinicaltrials.gov/ct2/show/NCT03661879. Accessed 4 Jun 2021
Nunez DJ, Bush MA, Collins DA, McMullen SL, Gillmor D, Apseloff G, Atiee G, Corsino L, Morrow L, Feldman PL (2014) Gut hormone pharmacology of a novel GPR119 agonist (GSK1292263), metformin, and sitagliptin in type 2 diabetes mellitus: results from two randomized studies. PLoS One 9:e92494. https://doi.org/10.1371/journal.pone.0092494
Nunez DJ, Yao X, Lin J, Walker A, Zuo P, Webster L, Krug-Gourley S, Zamek-Gliszczynski MJ, Gillmor DS, Johnson SL (2016) Glucose and lipid effects of the ileal apical sodium-dependent bile acid transporter inhibitor GSK2330672: double-blind randomized trials with type 2 diabetes subjects taking metformin. Diabetes Obes Metab 18:654–662. https://doi.org/10.1111/dom.12656
Otieno MA, Snoeys J, Lam W, Ghosh A, Player MR, Pocai A, Salter R, Simic D, Skaggs H, Singh B, Lim HK (2018) Fasiglifam (TAK-875): mechanistic investigation and retrospective identification of hazards for drug induced liver injury. Toxicol Sci 163:374–384. https://doi.org/10.1093/toxsci/kfx040
Pais R, Gribble FM, Reimann F (2016) Signalling pathways involved in the detection of peptones by murine small intestinal enteroendocrine L-cells. Peptides 77:9–15. https://doi.org/10.1016/J.PEPTIDES.2015.07.019
Parker HE, Adriaenssens A, Rogers G, Richards P, Koepsell H, Reimann F, Gribble FM (2012a) Predominant role of active versus facilitative glucose transport for glucagon-like peptide-1 secretion. Diabetologia 55:2445–2455. https://doi.org/10.1007/s00125-012-2585-2
Parker HE, Wallis K, le Roux CW, Wong KY, Reimann F, Gribble FM (2012b) Molecular mechanisms underlying bile acid-stimulated glucagon-like peptide-1 secretion. Br J Pharmacol 165:414–423. https://doi.org/10.1111/j.1476-5381.2011.01561.x
Peterli R, Steinert RE, Woelnerhanssen B, Peters T, Christoffel-Courtin C, Gass M, Kern B, von Fluee M, Beglinger C (2012) Metabolic and hormonal changes after laparoscopic Roux-en-Y gastric bypass and sleeve gastrectomy: a randomized, prospective trial. Obes Surg 22:740–748. https://doi.org/10.1007/s11695-012-0622-3
Petersen N, Reimann F, Bartfeld S, Farin HF, Ringnalda FC, Vries RG, van den Brink S, Clevers H, Gribble FM, de Koning EJ (2014) Generation of L cells in mouse and human small intestine organoids. Diabetes 63:410–420. https://doi.org/10.2337/db13-0991
Petersen N, Reimann F, van Es JH, van den Berg BM, Kroone C, Pais R, Jansen E, Clevers H, Gribble FM, de Koning EJ (2015) Targeting development of incretin-producing cells increases insulin secretion. J Clin Invest 125:379–385. https://doi.org/10.1172/JCI75838
Petersen N, Frimurer TM, Terndrup Pedersen M, Egerod KL, Wewer Albrechtsen NJ, Holst JJ, Grapin-Botton A, Jensen KB, Schwartz TW (2018) Inhibiting RHOA signaling in mice increases glucose tolerance and numbers of enteroendocrine and other secretory cells in the intestine. Gastroenterology 155:1164–1176.e2. https://doi.org/10.1053/j.gastro.2018.06.039
Potthoff MJ, Potts A, He T, Duarte JA, Taussig R, Mangelsdorf DJ, Kliewer SA, Burgess SC (2013) Colesevelam suppresses hepatic glycogenolysis by TGR5-mediated induction of GLP-1 action in DIO mice. Am J Physiol Gastrointest Liver Physiol 304:G371–G380. https://doi.org/10.1152/ajpgi.00400.2012
Powell DR, Smith MG, Doree DD, Harris AL, Greer J, DaCosta CM, Thompson A, Jeter-Jones S, Xiong W, Carson KG, Goodwin NC, Harrison BA, Rawlins DB, Strobel ED, Gopinathan S, Wilson A, Mseeh F, Zambrowicz B, Ding ZM (2017) LX2761, a sodium/glucose cotransporter 1 inhibitor restricted to the intestine, improves glycemic control in mice. J Pharmacol Exp Ther 362:85–97. https://doi.org/10.1124/jpet.117.240820
Rangwala SM, D'Aquino K, Zhang YM, Bader L, Edwards W, Zheng S, Eckardt A, Lacombe A, Pick R, Moreno V, Kang L, Jian W, Arnoult E, Case M, Jenkinson C, Chi E, Swanson RV, Kievit P, Grove K, Macielag M, Erion MD, SinhaRoy R, Leonard JN (2019) A long-acting PYY3-36 analog mediates robust anorectic efficacy with minimal emesis in nonhuman primates. Cell Metab 29:837–843.e5. https://doi.org/10.1016/j.cmet.2019.01.017
Rao A, Kosters A, Mells JE, Zhang W, Setchell KD, Amanso AM, Wynn GM, Xu T, Keller BT, Yin H, Banton S, Jones DP, Wu H, Dawson PA, Karpen SJ (2016) Inhibition of ileal bile acid uptake protects against nonalcoholic fatty liver disease in high-fat diet-fed mice. Sci Transl Med 8:357ra122. https://doi.org/10.1126/scitranslmed.aaf4823
Rhee NA, Wahlgren CD, Pedersen J, Mortensen B, Langholz E, Wandall EP, Friis SU, Vilmann P, Paulsen SJ, Kristiansen VB, Jelsing J, Dalboge LS, Poulsen SS, Holst JJ, Vilsboll T, Knop FK (2015) Effect of Roux-en-Y gastric bypass on the distribution and hormone expression of small-intestinal enteroendocrine cells in obese patients with type 2 diabetes. Diabetologia 58:2254–2258. https://doi.org/10.1007/s00125-015-3696-3
Roberts GP, Larraufie P, Richards P, Kay RG, Galvin SG, Miedzybrodzka EL, Leiter A, Li HJ, Glass LL, Ma MKL, Lam B, Yeo GSH, Scharfmann R, Chiarugi D, Hardwick RH, Reimann F, Gribble FM (2019) Comparison of human and murine enteroendocrine cells by transcriptomic and peptidomic profiling. Diabetes 68:1062–1072. https://doi.org/10.2337/db18-0883
Rudling M, Camilleri M, Graffner H, Holst JJ, Rikner L (2015) Specific inhibition of bile acid transport alters plasma lipids and GLP-1. BMC Cardiovasc Disord 15:75. https://doi.org/10.1186/s12872-015-0070-9
Ryan DH (2021) Semaglutide for obesity: four STEPs forward, but more to come. Lancet Diabetes Endocrinol 9:252–254. https://doi.org/10.1016/S2213-8587(21)00081-4
Ryan GJ, Jobe LJ, Martin R (2005) Pramlintide in the treatment of type 1 and type 2 diabetes mellitus. Clin Ther 27:1500–1512. https://doi.org/10.1016/j.clinthera.2005.10.009
Salehi M, Gastaldelli A, D'Alessio DA (2014) Blockade of glucagon-like peptide 1 receptor corrects postprandial hypoglycemia after gastric bypass. Gastroenterology 146:669–680.e2. https://doi.org/10.1053/j.gastro.2013.11.044
Samocha-Bonet D, Wong O, Synnott EL, Piyaratna N, Douglas A, Gribble FM, Holst JJ, Chisholm DJ, Greenfield JR (2011) Glutamine reduces postprandial glycemia and augments the glucagon-like peptide-1 response in type 2 diabetes patients. J Nutr 141:1233–1238. https://doi.org/10.3945/jn.111.139824
Sankyo D (2018) 12-Week study of DS-8500a in subjects with type 2 diabetes mellitus on metformin
Sanofi (2018) Form 20-F. https://www.sanofi.com/-/media/Project/One-Sanofi-Web/Websites/Global/Sanofi-COM/Home/common/docs/investors/Sanofi-20-F-2018-EN-PDF-e-accessible_02.pdf?la=en&hash=0E0CB0122FEDA3881B3857221DB9345A. Accessed 24 Jun 2021
Schiavon M, Visentin R, Gobel B, Riz M, Cobelli C, Klabunde T, Dalla Man C (2021) Improved postprandial glucose metabolism in type 2 diabetes by the dual glucagon-like peptide-1/glucagon receptor agonist SAR425899 in comparison with liraglutide. Diabetes Obes Metab. https://doi.org/10.1111/dom.14394
Schmidt JB, Gregersen NT, Pedersen SD, Arentoft JL, Ritz C, Schwartz TW, Holst JJ, Astrup A, Sjodin A (2014) Effects of PYY3-36 and GLP-1 on energy intake, energy expenditure, and appetite in overweight men. Am J Physiol Endocrinol Metab 306:E1248–E1256. https://doi.org/10.1152/ajpendo.00569.2013
Seifarth C, Bergmann J, Holst JJ, Ritzel R, Schmiegel W, Nauck MA (1998) Prolonged and enhanced secretion of glucagon-like peptide 1 (7-36 amide) after oral sucrose due to alpha-glucosidase inhibition (acarbose) in type 2 diabetic patients. Diabet Med 15:485–491. https://doi.org/10.1002/(SICI)1096-9136(199806)15:6<485::AID-DIA610>3.0.CO;2-Y
Sorensen KV, Korfitzen SS, Kaspersen MH, Ulven ER, Ekberg JH, Bauer-Brandl A, Ulven T, Hojlund K (2021) Acute effects of delayed-release hydrolyzed pine nut oil on glucose tolerance, incretins, ghrelin and appetite in healthy humans. Clin Nutr 40:2169–2179. https://doi.org/10.1016/j.clnu.2020.09.043
Sprecher U, Mohr P, Martin RE, Maerki HP, Sanchez RA, Binggeli A, Kunnecke B, Christ AD (2010) Novel, non-peptidic somatostatin receptor subtype 5 antagonists improve glucose tolerance in rodents. Regul Pept 159:19–27. https://doi.org/10.1016/j.regpep.2009.09.006
Svane MS, Jorgensen NB, Bojsen-Moller KN, Dirksen C, Nielsen S, Kristiansen VB, Torang S, Wewer Albrechtsen NJ, Rehfeld JF, Hartmann B, Madsbad S, Holst JJ (2016) Peptide YY and glucagon-like peptide-1 contribute to decreased food intake after Roux-en-Y gastric bypass surgery. Int J Obes 40:1699–1706. https://doi.org/10.1038/ijo.2016.121
Talsania T, Anini Y, Siu S, Drucker DJ, Brubaker PL (2005) Peripheral exendin-4 and peptide YY(3-36) synergistically reduce food intake through different mechanisms in mice. Endocrinology 146:3748–3756. https://doi.org/10.1210/en.2005-0473
Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, Macchiarulo A, Yamamoto H, Mataki C, Pruzanski M, Pellicciari R, Auwerx J, Schoonjans K (2009) TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab 10:167–177. https://doi.org/10.1016/j.cmet.2009.08.001
Tolhurst G, Heffron H, Lam YS, Parker HE, Habib AM, Diakogiannaki E, Cameron J, Grosse J, Reimann F, Gribble FM (2012) Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes 61:364–371. https://doi.org/10.2337/db11-1019
Trevaskis JL, Sun C, Athanacio J, D'Souza L, Samant M, Tatarkiewicz K, Griffin PS, Wittmer C, Wang Y, Teng CH, Forood B, Parkes DG, Roth JD (2015) Synergistic metabolic benefits of an exenatide analogue and cholecystokinin in diet-induced obese and leptin-deficient rodents. Diabetes Obes Metab 17:61–73. https://doi.org/10.1111/dom.12390
Tsakmaki A, Fonseca Pedro P, Pavlidis P, Hayee BH, Bewick GA (2020) ISX-9 manipulates endocrine progenitor fate revealing conserved intestinal lineages in mouse and human organoids. Mol Metab 34:157–173. https://doi.org/10.1016/j.molmet.2020.01.012
Ueno H, Ito R, Abe SI, Ookawara M, Miyashita H, Ogino H, Miyamoto Y, Yoshihara T, Kobayashi A, Tsujihata Y, Takeuchi K, Watanabe M, Yamada Y, Maekawa T, Nishigaki N, Moritoh Y (2019) SCO-267, a GPR40 full agonist, improves glycemic and body weight control in rat models of diabetes and obesity. J Pharmacol Exp Ther 370:172–181. https://doi.org/10.1124/jpet.118.255885
Unniappan S, McIntosh CH, Demuth HU, Heiser U, Wolf R, Kieffer TJ (2006) Effects of dipeptidyl peptidase IV on the satiety actions of peptide YY. Diabetologia 49:1915–1923. https://doi.org/10.1007/s00125-006-0310-8
van Nierop FS, Scheltema MJ, Eggink HM, Pols TW, Sonne DP, Knop FK, Soeters MR (2017) Clinical relevance of the bile acid receptor TGR5 in metabolism. Lancet Diabetes Endocrinol 5:224–233. https://doi.org/10.1016/S2213-8587(16)30155-3
West JA, Tsakmaki A, Ghosh SS, Parkes DG, Gronlund RV, Pedersen PJ, Maggs D, Rajagopalan H, Bewick GA (2021) Chronic peptide-based GIP receptor inhibition exhibits modest glucose metabolic changes in mice when administered either alone or combined with GLP-1 agonism. PLoS One 16:e0249239. https://doi.org/10.1371/journal.pone.0249239
Wilding JPH, Batterham RL, Calanna S, Davies M, Van Gaal LF, Lingvay I, McGowan BM, Rosenstock J, Tran MTD, Wadden TA, Wharton S, Yokote K, Zeuthen N, Kushner RF, Group SS (2021) Once-weekly semaglutide in adults with overweight or obesity. N Engl J Med 384:989. https://doi.org/10.1056/NEJMoa2032183
Xu Y, Van Hul M, Suriano F, Preat V, Cani PD, Beloqui A (2020) Novel strategy for oral peptide delivery in incretin-based diabetes treatment. Gut 69:911–919. https://doi.org/10.1136/gutjnl-2019-319146
Yamada Y, Terauchi Y, Watada H, Nakatsuka Y, Shiosakai K, Washio T, Taguchi T (2018) Efficacy and safety of GPR119 agonist DS-8500a in Japanese patients with type 2 diabetes: a randomized, double-blind, placebo-controlled, 12-week study. Adv Ther 35:367–381. https://doi.org/10.1007/s12325-018-0668-2
Yousseif A, Emmanuel J, Karra E, Millet Q, Elkalaawy M, Jenkinson AD, Hashemi M, Adamo M, Finer N, Fiennes AG, Withers DJ, Batterham RL (2014) Differential effects of laparoscopic sleeve gastrectomy and laparoscopic gastric bypass on appetite, circulating acyl-ghrelin, peptide YY3-36 and active GLP-1 levels in non-diabetic humans. Obes Surg 24:241–252. https://doi.org/10.1007/s11695-013-1066-0
Zhang Q, Delessa CT, Augustin R, Bakhti M, Collden G, Drucker DJ, Feuchtinger A, Caceres CG, Grandl G, Harger A, Herzig S, Hofmann S, Holleman CL, Jastroch M, Keipert S, Kleinert M, Knerr PJ, Kulaj K, Legutko B, Lickert H, Liu X, Luippold G, Lutter D, Malogajski E, Medina MT, Mowery SA, Blutke A, Perez-Tilve D, Salinno C, Sehrer L, DiMarchi RD, Tschop MH, Stemmer K, Finan B, Wolfrum C, Muller TD (2021) The glucose-dependent insulinotropic polypeptide (GIP) regulates body weight and food intake via CNS-GIPR signaling. Cell Metab 33:833–844.e5. https://doi.org/10.1016/j.cmet.2021.01.015
Zinman B, Aroda VR, Buse JB, Cariou B, Harris SB, Hoff ST, Pedersen KB, Tarp-Johansen MJ, Araki E, Investigators P (2019) Efficacy, safety, and tolerability of oral semaglutide versus placebo added to insulin with or without metformin in patients with type 2 diabetes: the PIONEER 8 trial. Diabetes Care 42:2262–2271. https://doi.org/10.2337/dc19-0898
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Miedzybrodzka, E.L., Gribble, F.M., Reimann, F. (2022). Targeting the Enteroendocrine System for Treatment of Obesity. In: Eckel, J., Clément, K. (eds) From Obesity to Diabetes. Handbook of Experimental Pharmacology, vol 274. Springer, Cham. https://doi.org/10.1007/164_2022_583
Download citation
DOI: https://doi.org/10.1007/164_2022_583
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-99994-0
Online ISBN: 978-3-030-99995-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)