
Abstract
Multiple sclerosis (MS) is an autoimmune disease of the central nervous system (CNS) characterized by peripheral immune cell infiltration into the brain and spinal cord, demyelination, glial cell activation, and neuronal damage. Currently there is no cure for MS, however, available disease-modifying agents minimize inflammation in the CNS by various mechanisms. Approved drugs lessen severity of the disease and delay disease progression, however, they are still suboptimal as patients experience adverse effects and varying efficacies. Additionally, there is only one disease-modifying therapy available for the more debilitating, progressive form of MS. This chapter focuses on the presently-available therapeutics and, importantly, the future directions of MS therapy based on preclinical studies and early clinical trials. Immunosuppression in other neurological disorders including neuromyelitis optica spectrum disorders, myasthenia gravis, and Guillain-Barré syndrome is also discussed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Autoimmunity
- Disease-modifying therapies
- Guillain-Barré syndrome
- Immunosuppression
- Multiple sclerosis
- Myasthenia gravis
- Neuromyelitis optica
1 Introduction
Multiple sclerosis (MS) is a chronic, demyelinating autoimmune disease of the central nervous system (CNS), affecting approximately 2.5 million people worldwide (Reich et al. 2018; Trapp and Nave 2008). The condition affects females more often than males (Reich et al. 2018; Dendrou et al. 2015) and though the etiology is still poorly understood, it is thought that both genetic and environmental factors play a causative role in the development of MS (Reich et al. 2018; Hauser and Oksenberg 2006). Clinical symptoms of the disease include disturbances in motor function, vision, and speech, fatigue, acute/chronic pain, and in severe cases, paralysis and cognitive impairment. Symptoms are caused by multifocal lesions in the brain and spinal cord that consist of inflammation, demyelination, blood-brain barrier (BBB) breakdown, peripheral immune cell infiltration, reactive gliosis, loss of oligodendrocytes, and axonal degeneration (Dutta and Trapp 2011; Trapp and Nave 2008).
MS is a heterogeneous condition consisting of different presentations and varying disease courses. Despite this, MS has been broadly categorized into subtypes: approximately 85% of patients are diagnosed with relapsing-remitting MS (RRMS) where symptomatic flare-ups, or relapses, are followed by periods of varying degrees of recovery. In majority of RRMS cases (~80%), patients progress to experience gradual worsening of relapses and fewer periods of recovery, termed secondary progressive MS (SPMS). A smaller fraction of patients experience progressing symptoms from the time of disease onset, a pattern recognized as primary progressive MS (PPMS). And yet another small subset of patients experience benign MS, where relapses are mild compared to RRMS and SPMS does not develop (Trapp and Nave 2008; Ransohoff et al. 2015; Hemmer et al. 2002).
As expected by the heterogeneity of its presentation and various forms, MS is defined by pathological alterations involving numerous cells types, both immune and non-immune. The primary pathological hallmarks of MS are areas of demyelination (referred to as “plaques” or “lesions”) in the white and gray matter of the brain and/or spinal cord. Demyelination is mediated by both innate and adaptive immune cells. Though the CNS is normally considered an “immune-privileged” site due to the multicellular vascular blood-brain barrier (BBB), disruption of the BBB is apparent in all clinical subtypes of MS. This disruption allows peripheral immune cells to infiltrate the brain/spinal cord tissue.
T and B lymphocytes seem to be selectively recruited to the CNS by myelin autoantigens in MS and various hypotheses exist as to what triggers this recruitment. A CNS intrinsic model hypothesizes that events within the CNS result in the release of autoantigens into the periphery. On the other hand, an extrinsic model suggests that a peripheral insult, such as a system infection, leads to an aberrant immune response against myelin (Thompson et al. 2018).
Historically, MS has been considered a primarily T-cell-mediated disease with both CD4+ and CD8+ T cells present in MS lesions. CD4+ T helper cells typically predominate in acute lesions, whereas CD8+ cytotoxic T cells are found in chronic plaques (Chitnis 2007). B cells, on the other hand, are only recently becoming recognized as drivers of MS pathology. B cells produce antibodies that recognize various myelin epitopes and can also serve as antigen-presenting cells (APCs), communicating with T cells (Sospedra 2018). B cells can polarize T helper cells by secreting cytokines. Specifically, B-cell production of interleukin-6 (IL-6) seems to drive the autoimmune process by inhibiting the conversion of conventional T cells into regulatory T cells (Tregs) which are capable of immune suppression (Korn et al. 2008).
Cells of the innate immune system also infiltrate the CNS. Studies in MS animal models have implicated blood-derived monocytes as drivers of MS pathology, though it has been difficult to dissect their roles compared to the CNS resident innate immune cells, microglia. Both cells have been characterized to possess both harmful and beneficial functions as they can both secrete inflammatory cytokines and chemokines, but they can also produce growth factors and phagocytose myelin debris, a major obstacle to remyelination (Kotter et al. 2006). Studies on MS brain samples have shown that activated microglia in plaque regions express high levels of major histocompatibility complex class II (MHC II) molecules, suggestive of increased and active antigen presentation, stimulating the adaptive immune system and worsening the disease process (Boyle and McGeer 1990; Zhang et al. 2011; Raivich and Banati 2004). Other studies in animal models have suggested that the infiltrating monocyte-derived macrophages are the main drivers of pathology (Ajami et al. 2011; Yamasaki et al. 2014).
Although MS is probably the most well-recognized autoimmune disease of the CNS, there are several other neurological conditions with an autoimmune component, requiring pharmacological immunosuppression. Here, we discuss both current strategies to dampen the autoimmune response in MS as well as other neurological conditions, including neuromyelitis optica spectrum disorders (NMOSD), myasthenia gravis (MG), and Guillain-Barré syndrome. Importantly, we highlight potential future immunosuppressive therapies that may further improve the clinical treatment of MS.
2 Current Strategies to Promote Immunosuppression in Multiple Sclerosis
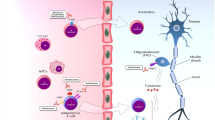
Therapeutic management of MS currently relies on immunomodulation to dampen the autoimmune response occurring in the CNS. Available disease-modifying therapies (DMTs) can be sorted into broad classifications based upon their mechanism of immunosuppression: (1) pleiotropic immunomodulators, (2) drugs interfering with DNA synthesis and repair, (3) reagents that sequester peripheral leukocytes, and (4) reagents that deplete immune cells. There are also non-DMTs that are commonly used to control relapses in RRMS. Corticosteroids, such as high-dose intravenous methylprednisolone, are the first line of treatment for acute symptomatic exacerbations. A recent study reported that oral administration of high-dose methylprednisolone was similar in efficacy and safety compared to the intravenous route (Le Page et al. 2015). Orally administered medications are favorable not only for patient convenience, but also because phobia of needles, impaired dexterity, and reactions at injection sites often result in poor patient compliance (Mohr et al. 2001). In this section, we will discuss currently approved DMTs based upon their mechanisms of immune suppression (Fig. 1).

Targets of currently approved DMTs (indicated in red) in the pathological setting of MS. DMTs with pleiotropic mechanisms are indicated in gray box
2.1 Pleiotropic Immunosuppressants
The first DMT, recombinant interferon-β (IFN-β), is a pleiotropic drug and remains a leading therapeutic option for RRMS since its approval by the United States Food and Drug Administration (FDA) in 1993. Its availability to patients marked a significant milestone in MS therapy as it was the first time the disease was viewed as treatable (Ransohoff et al. 2015). Different forms of the drug are now available including IFN-β1b (Betaseron, Betaferon, Extavia) and IFN-β1a (Avonex, Rebif, Plegridy), though IFN-β1b was the first to be studied and approved. In the first multicenter study of 372 RRMS patients, IFN-β1b was shown to reduce annual relapse rate by ~30% (Paty and Li 1993). Recently, an 11-year clinical study showed that early treatment with IFN-β1b in patients with clinically isolated syndrome (CIS; suggestive of a first MS attack) resulted in long-term benefits (Hartung et al. 2019). The recombinant cytokine binds the heterodimeric, multi-subunit IFN-β receptor (IFNAR1 and IFNAR2), resulting in Janus Activated Kinase-Signal Transducer and Activator of Transcription (JAK-STAT) signaling and its pleiotropic effects are the result of transcriptional effects on hundreds of genes (Hojati et al. 2016). The most prominent immunosuppressive actions of IFN-β include inhibition of T-cell activation through decreased expression of MHCII and co-stimulatory molecules, increased apoptosis of autoreactive T cells, and reduction in the stimulatory capacity of B cells (Dhib-Jalbut and Marks 2010). A cytokine shift has also been observed upon IFN-β treatment, inhibiting Th1 pro-inflammatory cytokines and promoting release of Th2 anti-inflammatory cytokines (Ersoy et al. 2005).
There are patients, however, who do not respond well to IFN-β and exhibit either severe side effects or no improvement in disease activity. Another pharmacological option became available in 1997 with the approval of glatiramer acetate (GA; Copaxone), a synthetic copolymer of amino acids analogous to an epitope of myelin basic protein (MBP). Interestingly, GA was discovered when Teitelbaum and colleagues sought to produce a synthetic antigen capable of inducing experimental autoimmune encephalomyelitis (EAE), the primary animal model of MS. Surprisingly, rather than inducing disease, GA protected against EAE induction (Teitelbaum et al. 1971). In clinical trials, GA reduced the relapse rate and was relatively well-tolerated in humans (Johnson et al. 1995; Comi et al. 2009). It also displayed comparable efficacy to IFN-β formulations. A major mechanism of action of GA is the induction of apoptosis in CD4+ T cells, and a recent study in RRMS patients suggests that this is a biomarker of optimal treatment response (Boziki et al. 2019). GA was shown to increase the number of anti-inflammatory monocytes and immunosuppressive Tregs, maintaining these effects over a decade of GA administration (Spadaro et al. 2017).
Dimethyl fumarate (DMF, Tecfidera), another pleiotropic drug, was approved as a first-line treatment for RRMS in 2013. DMF activates the transcription factor, nuclear factor (erythroid-derived 2)-like 2 (Nrf2), which is responsible for maintaining cellular redox homeostasis. When transported to the nucleus, Nrf2 induces expression of antioxidants and detoxifying enzymes (Ma 2013). DMF also modulates Nrf2-independent pathways. For instance, the agent suppresses NF-κB signaling, resulting in the reduction of inflammatory cytokines and induction of Th2, anti-inflammatory phenotypes (Gillard et al. 2015). Importantly, a recent study reported persistent changes in both the innate and adaptive immune system in MS patients after 12 months of DMF treatment, observing a decrease in effector memory T cells, memory B cells, and expression of antigen presentation molecules (Montes Diaz et al. 2018).
2.2 Drugs Interfering with DNA Synthesis/Repair
Mitoxantrone (Novantrone) was initially approved as an antineoplastic agent as it globally disrupts DNA synthesis through inhibition of type II topoisomerase (Shenkenberg and Von Hoff 1986). Mitoxantrone is generally immunosuppressive, and is only prescribed in cases of rapidly worsening MS. Although a multicenter study of patients with severe and worsening RRMS or progressive MS showed that mitoxantrone did reduce progression of disability (Hartung et al. 2002), its use in MS has dramatically decreased due to severe side effects, such as cardiac toxicity and acute leukemia (Capobianco et al. 2008), and the approval of less dangerous medications.
Teriflunomide (Aubagio) is an oral inhibitor of dihydroorotate dehydrogenase (DHODH), a mitochondrial enzyme necessary for de novo pyrimidine synthesis. Inhibition of this enzyme limits availability of pyrimidines in proliferating T and B cells, reducing the number of autoreactive lymphocytes available to cross the BBB (Claussen and Korn 2012). Three large phase III trials showed that 7–14 mg of teriflunomide decreased annual relapse rates and MRI disease activity, which resulted in the approval of the drug in 2004 (O’Connor et al. 2011). A more recent 9-year follow-up study showed that long-term treatment remains efficacious and is well-tolerated in patients (O’Connor et al. 2016).
Cladribine (Mavenclad), a synthetic chlorinated deoxyadenosine analog, is the most recent drug approved by the FDA for MS. Cladribine is taken up by cells, and undergoes several phosphorylation steps to produce the active compound, 2-chlorodeoxyadenosis 5′-triphosphate (2-CdATP). 5′-nucleotidases in most cells degrade 2-CdATP, however, lymphocytes have lower levels of these enzymes and higher levels of deoxycytidine kinase (DCK), the enzyme responsible for cladribine phosphorylation. This ultimately results in intracellular accumulation of 2-CdATP selectively in lymphocytes, and the active compound becomes incorporated into DNA, leading to strand breaks and cell death (Leist and Weissert 2011; Baker et al. 2019). In comparison with monoclonal antibodies that deplete B cells, such as ocrelizumab and rituximab, cladribine’s mode of action results in a more gradual depletion (Montalban et al. 2017; Baker et al. 2019). A recent study showed that after 20 days of oral treatment, CD19+ B cells and CD8+ T cells return to baseline levels, and patients maintain no clinical or MRI disease activity. Further, monocyte and neutrophil numbers remain intact resulting in less risk of opportunistic infections (Comi et al. 2019).
2.3 Reagents That Sequester Peripheral Leukocytes
Fingolimod (FTY720; Gilenya), which reduces CNS inflammation by limiting lymphocytes in the periphery, was the first oral medication for RRMS patients. Approved by the FDA in 2010, fingolimod is a sphingosine-1-phosphate (S1P) receptor antagonist that prevents T- and B-cell egress from lymph nodes, reducing the number of autoreactive lymphocytes in the CNS. A phase III study reported that oral treatment with fingolimod for 12 months was superior to intramuscular IFN-β1a in terms of annualized relapse rate and MRI disease activity (Cohen et al. 2010).
Natalizumab (Tysabri) is a humanized monoclonal antibody against the α4 subunit of the very late antigen 4 (VLA4) integrin expressed on leukocytes. Blockage of this cell adhesion protein functions to prevent lymphocyte migration into the CNS as it blocks interaction with vascular-cell adhesion molecule 1 (VCAM-1) on vascular endothelial cells in the brain and spinal cord (Ransahoff 2007). Natalizumab was studied as both a monotherapy and an IFN-β therapy. Both phase III clinical trials took place over the course of 2 years and included only RRMS patients. As a monotherapy, natalizumab reduced relapse rate and gadolinium-enhancing lesions on MRI at year 2 by 92% (Polman et al. 2006). When natalizumab was administered to patients on IFN-β, who had at least one relapse during the past year of treatment, the combination of the drugs was observed to be significantly more effective than interferon alone (Rudick et al. 2006). The use of natalizumab is limited by the occurrence of progressive multifocal leukoencephalopathy (PML), a fatal brain infection, and current studies are attempting to establish biomarkers to predict the risk of PML in MS patients (Schwab et al. 2013, 2016).
2.4 Reagents Depleting Immune Cells
Ocrelizumab (Ocrevus) is an anti-CD20 antibody, acting to deplete CD20-expressing B cells. The approval of this agent was groundbreaking in the field of MS therapeutics as it was the first drug to show efficacy for patients with PPMS (Mulero et al. 2018). A phase III placebo-controlled trial of 732 PPMS patients reported that those receiving ocrelizumab displayed lower rates of progression (assessed clinically and by MRI) compared to the placebo group (Montalban et al. 2017). The remarkable results of the anti-CD20 therapy have renewed interest in the role of B cells in MS pathology, as MS has historically been considered a primarily T-cell-mediated disease.
Alemtuzumab (Lemtrada), a humanized monoclonal antibody targeting CD52 on lymphocytes, monocytes, granulocytes, and natural killer (NK) cells induces rapid lymphopenia through antibody-dependent cellular cytotoxicity (ADCC). Clinical studies showed that infusion of alemtuzumab decreased annualized relapse rate, reduced disability progression, and reduced MRI disease activity. Further, it was observed to be superior to IFN-β1a therapy (Coles et al. 2012; Cohen et al. 2012). The most common adverse effect is secondary autoimmunity, most typically involving the thyroid gland. A long-term follow-up study confirmed that alemtuzumab stabilizes disease in patients with highly active RRMS (Tuohy et al. 2015).
Daclizumab (Zinbryta) is a humanized monoclonal antibody against the CD25 subunit of the interleukin-2 (IL-2) receptor, highly expressed on activated T cells. This results in functional impairment of the T cells. Daclizumab treatment results in a decrease in circulating CD4+ and CD8+ T cells and expansion of CD56bright natural killer (NK) cells, which is considered an immunoregulatory NK cell population due to cytokine profiles and expansion during states of immune tolerance (Bielekova et al. 2006). Daclizumab approved in 2016 is prescribed only to patients who are refractory to at least two first-line treatments (Baldassari and Rose 2017). Interestingly, a phase II study that added daclizumab therapy on to IFN-β treatment found that the combination may more effectively reduce disease activity compared to IFN-β alone (Wynn et al. 2010).
3 Looking Ahead: The Future of Immunosuppressive Therapies for Multiple Sclerosis
Though there has been truly amazing progress in the field of MS therapeutics over the past decade, the limitations of currently available agents justify continued research efforts to improve treatment options for patients. Available immunotherapies are variable in their efficacies, often produce adverse effects, and ultimately are unable to prevent disease progression. Although there is an abundance of preclinical, and some clinical, focus on addressing these limitations by studying agents capable of promoting remyelination and repair, here, we will focus solely on innovative approaches to improve therapies that modulate the immune system in MS.
3.1 Targeting B Cells
Recently, B cells have gained attention as an exciting and potentially more effective therapeutic target in various subtypes of MS due to the success of ocrelizumab, and other B-cell-targeted antibodies in clinical trials (namely, rituximab and ofatumumab). Other agents that modulate B cells by various mechanisms are likely to enter the clinic and be approved in the coming years.
Early clinical studies have reported positive results in RRMS patients treated with Bruton’s tyrosine kinase (BTK) inhibitors. BTK, a non-receptor tyrosine kinase, regulates B-cell function, playing a central role in B-cell receptor (BCR) signaling. BTK signaling pathways also modulates myeloid cells. BTK inhibitors have been available in recent years for the treatment of B-cell leukemias/lymphomas (Liang et al. 2018). Now, newer and more selective inhibitors have been developed and are currently being investigated in not only B-cell malignancies, but also in autoimmune settings, such as rheumatoid arthritis and MS (Zhang et al. 2018). A phase II clinical trial reported that after 24 weeks of once daily oral treatment with 75-mg of the BTK inhibitor, evobrutinib, RRMS patients displayed decreased gadolinium-enhancing lesions on T1-weighted MRI compared to patients receiving placebo (Montalban et al. 2019). A phase III study has been posted to compare evobrutinib’s effectiveness to the current first-line treatment, IFN-β1a (NCT04032171).
Another B-cell-directed therapeutic target under investigation is B-cell-activated factor (BAFF). BAFF is a member of the tumor necrosis factor family which promotes B-cell development and survival. It has been observed to be elevated in the cerebrospinal fluid (CSF) of MS patients (Ragheb et al. 2011) as well as accumulate in inflammatory demyelinating brain lesions (Krumbholz et al. 2005). A humanized recombinant fusion protein, Atacicept, was developed to block both BAFF and a proliferation-inducing ligand (APRIL), which is also involved in B-cell differentiation and maturation signaling. After preclinical work showing a decrease in mature B cells (Gross et al. 2001), a phase II trial also showed that Atacicept treatment did reduce serum immunoglobulin and number of circulating mature B cells in RRMS patients, however, there was an unexpected increase in relapses. The cellular and symptomatic effects did revert to that of the placebo group after discontinuation of the drug, illustrating reversibility of the mechanism (Kappos et al. 2014). VAY736, a humanized monoclonal antibody against one of the receptors for BAFF (BAFF-R) has also been evaluated in RRMS patients in a phase II trial, however, results are not yet posted (NCT02038049).
3.2 Stem Cell Therapies
Hematopoietic stem cells (HSCs) are the primary stem cell population of the bone marrow, capable of giving rise to all types of blood cells. Hematopoietic stem cell transplantation (HSCT) has long been used as a method to treat hematological malignancies, but only in the early 1990s was it considered for use in MS patients after pivotal preclinical studies (Karussis et al. 1992, 1993). It is important to note that stem cell transplant is a high-risk procedure with aggressive immunoablation to extinguish pathogenic immune cells and “reset” the immune system with only HSCs. Typically, a patient’s own HSCs are employed (autologous HSCT; aHSCT) (Karussis and Petrou 2018).
A multicenter phase II trial showed that aHSCT in MS patients with poor prognosis (both RRMS and SPMS subtypes) led to long-lasting remission in the majority of patients with no DMT regimen. Further, there was significant neurological improvement as the rate of brain atrophy decreased to that of healthy aging controls (Atkins et al. 2016). A larger-scale study also reported that approximately half of the patients undergoing HSCT did not exhibit neurological progression 5 years post-transplant. Successful outcomes were associated with younger age, an RRMS subtype, and fewer previous immunotherapies (Muraro et al. 2017). Though small, a recent study in Sweden showed that five out of ten patients exhibited sustained remission 10 years after aHSCT, and the investigators suggested that MS was “resolved” (characterized by normalized intrathecal IgG production and CSF neurofilament light levels) in three out of the five patients (Tolf et al. 2019). Though exciting results continue to be obtained, the risks of the procedure remain a concern and thus, development of new, safer protocols is required to consider this a standard therapy.
Mesenchymal stem cells (MSCs) are another cell therapy currently explored for severe cases of MS. MSCs are stromal precursor cells which, in the bone marrow, function to support hematopoiesis and display highly anti-inflammatory properties, inhibiting lymphocyte and APC function and modulating T-regulatory cells (Karampera et al. 2003; Corcione et al. 2006; Di Ianni et al. 2008; Beyth et al. 2005). In preclinical studies using the EAE model, MSC transplant therapy is reported to not only be anti-inflammatory, but also neuroprotective, supporting remyelination of damaged axons (Kassis et al. 2008; Zappia et al. 2005). In clinical studies of MS patients, bone marrow-derived MSC administration was observed to be generally well-tolerated, but small sample sizes limited conclusions concerning efficacy of the treatment (Yamout et al. 2010; Bonab et al. 2012). Recently, the Mesenchymal Stem cells for Multiple Sclerosis (MESEMS) study group published their protocol for a larger-scale phase I/II study that aims to evaluate the safety and activity of intravenous autologous bone marrow-derived MSCs in patients with RRMS, SPMS, and PPMS (Uccelli et al. 2019). Additionally, studies have evaluated the safety and efficacy MSC-derived neural progenitors, which were shown to be neurotrophic and immunoregulatory. A phase I trial of MSC-derived neural progenitors administered intrathecally to patients with progressive MS showed that the treatment was well-tolerated with only minor adverse events occurring. Further, evidence of clinical disability trended towards improvement following treatment (Harris et al. 2018).
4 Immunosuppressants for Other Neurologic Disorders
4.1 Neuromyelitis Optica Spectrum Disorders
Neuromyelitis optica spectrum disorders (NMOSD) is a group of relapsing neuroinflammatory diseases distinct from MS in its pathophysiology and thus, the approach to immunosuppression also differs (though there are some commonalities). Progression is rare in NMOSD, but relapses are very severe and characterized by complete vision loss and/or extreme motor/sensory dysfunctions as a result of inflammatory lesions formation in the spinal cord (Wingerchuk and Weinshenker 2003). NMOSD is typically distinguished from other CNS autoimmune disorders by the presence of an IgG autoantibody against the water channel, aquaporin 4 (AQP4) (Papadopoulos and Verkman 2012), though not all patients are anti-AQP4 positive. First-line therapy for acute relapse of NMOSD is comprised of corticosteroid treatment (typically high-dose intravenous methylprednisolone). Plasma exchange is the next option for progressive or refractory conditions (Kleiter et al. 2018). NMOSD relapses are disabling with patients rarely experiencing full recovery, therefore, at least 5 years of maintenance immunotherapy is standard, with the intent of preventing relapses and accumulation of disability (Patterson and Goglin 2017).
Chronic low-dose corticosteroids are one option to prevent NMOSD attacks, usually in combination with another immunosuppressive. However, long-term use of corticosteroids often results in adverse effects such as hyperglycemia, hypertension, and osteoporosis (Kleiter and Gold 2016). Azathioprine, a purine antagonist that acts to inhibit DNA synthesis, has been found to be effective in long-term treatment of NMOSD (either with or without prednisone), more so than steroid therapy alone (Costanzi et al. 2011; Mandler et al. 1998; Bichuetti et al. 2010). Another option is mycophenolate mofetil, a drug that is indicated for psoriasis and renal transplant rejection, but is also often employed in NMOSD as well. Mycophenolate mofetil is a prodrug, the active metabolite being mycophenolic acid, which inhibits lymphocyte proliferation by preventing guanosine nucleotide biosynthesis (Mealy et al. 2014; Jacob et al. 2009). Mitoxantrone, previously discussed above as a therapeutic option for MS, has also been observed to be beneficial, reducing the annualized relapse rate in the first year of treatment in patients with highly relapsing NMOSD (Kim et al. 2011b). Interestingly the first-line therapy for MS, IFN-β, as well as the DMTs natalizumab and fingolimod, exacerbate NMOSD (Shimizu et al. 2008; Kleiter et al. 2012; Min et al. 2012).
B-cell depletion has become an obvious therapeutic strategy due to the presence of AQP4 autoantibodies in majority of NMOSD patients. Cree et al. showed that six out of eight patients were relapse-free after one year of treatment with rituximab, an anti-CD20 antibody (Cree et al. 2005). Other studies have confirmed the safety and efficacy of rituximab treatment with a modified protocol; rather than the standard fixed maintenance therapy with rituximab every 6 months, the investigators only retreated with rituximab after determining whether the frequency of CD27+ memory B cells in peripheral blood of NMOSD patients exceeded 0.05% for the initial 2 years of treatment, and 0.1% thereafter. They observed a reduction in relapse rate and improvement of disability (Kim et al. 2011a, 2013). A more recent report assessed long-term (>7 years) treatment of NMOSD patients with the aforementioned treatment regimen. It was concluded that this long-term, modified approach was beneficial as no patients experienced serious side effects, there was a 97% reduction in annualized relapse rate compared to fixed treatment, memory B-cell population remained low, and unnecessary treatments with rituximab were avoided through the monitoring protocol (Kim et al. 2019).
Stem cell therapies are also under investigation for the treatment of severe, refractory NMOSD. A small study of two patients reported disappearance of anti-AQP4 antibodies, reduction of spinal cord lesions, and clinical remission 3-years post-allogeneic HSCT. Interestingly, the patients from this study had previously undergone aHSCT, indicating that allogeneic stem cell transplant may be more effective than autologous (Greco et al. 2014). A phase II/III trial is currently recruiting NMOSD patients to test an aggressive, investigational aHSCT procedure after conditioning with rituximab, cyclophosphamide, and antithymocyte globulin, a rabbit polyclonal antibody to deplete lymphocytes (NCT03829566).
4.2 Myasthenia Gravis
Myasthenia gravis (MG) is an autoimmune disease of the neuromuscular junction in which autoantibodies interfere with nerve-muscle communication. Patients typically test positive for anti-acetylcholine receptor (AChR) antibodies. Of those who are negative for AChR autoantibodies, ~40% will harbor antibodies for muscle-specific tyrosine kinase (MuSK) (Tandan et al. 2017). Autoimmune disruption in nerve-muscle conduction manifests as muscle fatigue and weakness in MG patients. As with MS and NMOSD, the initial treatment is typically corticosteroids to suppress inflammation; however, long-term use is limited by adverse effects (Gotterer and Li 2016) and most patients do require long-term immunosuppression to remain in remission.
Both azathioprine and mycophenolate mofetil are commonly prescribed to MG patients, as well as NMOSD, as previously discussed. An important 1998 randomized, double-blind trial compared prednisolone alone with prednisolone plus azathioprine in MG patients. Here, they found that the addition of azathioprine to corticosteroid treatment was able to reduce the maintenance dose of prednisolone, reduce side effects, and reduce relapses over the course of 3 years (Palace et al. 1998). Mycophenolate mofetil was first reported to be rapidly effective in a case study of a 26-year-old MG patient whose symptoms were previously difficult to manage with other immunosuppressants (Hauser et al. 1998). A few years after the publication of this case report, a retrospective study reported efficacy, but a more delayed onset of action of mycophenolate mofetil in MG patients. The investigators believe that since mycophenolate mofetil inhibits purine synthesis (preventing proliferation of lymphocytes), it does not kill pre-existing autoreactive lymphocytes, thus, the gradual death of the activated cells prior to treatment is what shows initial symptomatic improvement (Chaudhry et al. 2001).
Cyclosporine and tacrolimus (FK506), both calcineurin inhibitors which inhibit T-cell function by blocking the synthesis of interleukin-2 (IL-2) and interferon, are also often administered in MG cases as long-term immunosuppressants, allowing tapering/discontinuation of corticosteroids. However, cyclosporine use is often discontinued due to adverse effects, most often nephrotoxicity, that occur over time (Ciafaloni et al. 2000). Although tacrolimus is more well-tolerated in comparison with cyclosporine, there are still incidences of side effects (Nagaishi et al. 2008; Minami et al. 2011).
Eculizumab was recently approved by the FDA for MG after a phase III trial (REGAIN) that showed that, though the agent didn’t significantly improve the primary endpoint of MG-“Activities of Daily Living” Score, it did decrease exacerbations, need for rescue therapy, and hospital admissions. Eculizumab is a monoclonal antibody against the complement protein, C5, preventing formation of the terminal complement complex, C5b-9 (Howard et al. 2017).
Expectedly rituximab, as an off-label therapy, has also been observed to benefit patients with MG by depleting B cells and thus, decreasing levels of autoantibodies. A recent systematic retrospective analysis of the safety and efficacy of rituximab in MG patients reported that the agent was safe and effective for both AChR- and MuSK-positive MG patients, with a more robust response evident in the MuSK subset. Though, this study was unable to conclude what an optimal rituximab treatment regimen was comprised of due to the limitations associated with the reviewed case reports (Tandan et al. 2017). Another recent retrospective study assessed the long-term efficacy and safety of repeated treatments with low-dose rituximab in patients with severe, refractory MG. Here, it was reported that the repeated low-dose treatments, as guided by circulating CD19+ B-cell repopulation, was an effective therapy for difficult-to-manage MG (Choi et al. 2019). Further, a large nationwide study in Austria reported rituximab to be safe, rapidly efficacious, and provide the greatest benefits in MuSK-positive MG patients (Topakian et al. 2019). Rituximab is not without limitations, however, as severe adverse events, notably the development of PML, can occur.
4.3 Guillain-Barré Syndrome
Guillain-Barré syndrome (GBS) is an acute autoimmune disease resulting in demyelination of the peripheral nerves. It is characterized by immunoglobulin and complement-mediated attack on axons, as well as T cell and macrophage infiltration of peripheral nerves. Autoantibodies against gangliosides are often present in the serum of GBS patients and bind to Schwann cell surfaces, nodes of Ranvier, and peripheral axons (Ang et al. 2004). Patients experience rapid (on the scale of weeks), progressive weakness of the limbs, most often bilaterally, and with or without involvement of respiratory muscles. It is believed that GBS is caused by infection, which induces an aberrant immune response against the peripheral nerves (Hughes and Cornblath 2005). In particular, there is an abundance of evidence drawing an association between Campylobacter jejuni infection and the development of GBS (Ang et al. 2004). Though the majority of patients improve without immunotherapy, it is believed that early immunosuppression can reduce disease severity and facilitate a quick recovery. Plasma exchange was the first therapy to show efficacy in a 1985 randomized trial (Group 1985) and is now considered the gold standard treatment (Hughes and Cornblath 2005), with randomized clinical trials and large-scale studies supporting its use (Chevret et al. 2017). There have been a number of clinical studies showing that intravenous immunoglobulin (IVIG) is effective as well (Hughes et al. 2014; Van der Meche 1992). In contrast to the previously discussed neuroinflammatory diseases, corticosteroids are ineffective in GBS (Hughes et al. 2016). Unfortunately, treatments to completely prevent (or reverse) lingering disability are still lacking, and the development of improved therapies is critical.
5 Conclusion
This review highlights current as well as potential up-and-coming immunosuppressive strategies for neurologic conditions. The past decade has been truly remarkable in availability of novel immunomodulatory options for patients with MS and other neuroinflammatory diseases. Though a number of drugs are now approved for MS, there is still a critical need for the characterization and development of agents that are capable of suppressing the immune system while limiting adverse effects. Further, the largest unmet need in the field of MS therapeutics is modulating the immune system to halt or, more ideally, reverse disease progression. Additionally, as evident by the more limited therapies available for NMOSD, MG, and GBS, these conditions are all in need of more therapeutic options for patients who experience severe, refractory disease. Overall, however, current immunosuppressive regimens have enabled the treatment of these disabling neurologic autoimmune diseases, in most cases slowing progression and improving the patients’ quality of life. There is no doubt these strategies will continue to be refined, and new immunosuppressive approaches will be available in the future to enhance outcomes for these patients.
References
Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FM (2011) Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci 14(9):1142–1149. https://doi.org/10.1038/nn.2887
Ang CW, Jacobs BC, Laman JD (2004) The Guillain-Barre syndrome: a true case of molecular mimicry. Trends Immunol 25(2):61–66. https://doi.org/10.1016/j.it.2003.12.004
Atkins HL, Bowman M, Allan D, Anstee G, Arnold DL, Bar-Or A, Bence-Bruckler I, Birch P, Bredeson C, Chen J, Fergusson D, Halpenny M, Hamelin L, Huebsch L, Hutton B, Laneuville P, Lapierre Y, Lee H, Martin L, McDiarmid S, O'Connor P, Ramsay T, Sabloff M, Walker L, Freedman MS (2016) Immunoablation and autologous haemopoietic stem-cell transplantation for aggressive multiple sclerosis: a multicentre single-group phase 2 trial. Lancet 388(10044):576–585. https://doi.org/10.1016/s0140-6736(16)30169-6
Baker D, Pryce G, Herrod SS, Schmierer K (2019) Potential mechanisms of action related to the efficacy and safety of cladribine. Mult Scler Relat Disord 30:176–186. https://doi.org/10.1016/j.msard.2019.02.018
Baldassari LE, Rose JW (2017) Daclizumab: development, clinical trials, and practical aspects of use in multiple sclerosis. Neurotherapeutics 14(4):842–858. https://doi.org/10.1007/s13311-017-0553-8
Beyth S, Borovsky Z, Mevorach D, Liebergall M, Gazit Z, Aslan H, Galun E, Rachmilewitz J (2005) Human mesenchymal stem cells alter antigen-presenting cell maturation and induce T-cell unresponsiveness. Blood 105(5):2214–2219
Bichuetti DB, Lobato de Oliveira EM, Oliveira DM, de Souza NA, Gabbai AA (2010) Neuromyelitis optica treatment: analysis of 36 patients. Arch Neurol 67(9):1131–1136
Bielekova B, Catalfamo M, Reichert-Scrivner S, Packer A, Cerna M, Waldmann TA, McFarland H, Henkart PA, Martin R (2006) Regulatory CD56bright natural killer cells mediate immunomodulatory effects of IL-2Ra-targeted therapy (daclizumab) in multiple sclerosis. PNAS 103(15):5941–5946
Bonab MM, Sahraian MA, Aghhsaie A, Karvigh SA, Hosseinian SM, Nikbin B, Lotfi J, Khorramnia S, Motamed MR, Togha M, Harirchian MH, Moghadam NB, Alikhani K, Yadegari S, Jafarian S, Gheini MR (2012) Autologous mesenchymal stem cell therapy in progressive multiple sclerosis: an open label study. Curr Stem Cell Res Ther 7:407–414
Boyle EA, McGeer PL (1990) Cellular immune response in multiple sclerosis plaques. Am J Pathol 137(3):575–584
Boziki M, Lagoudaki R, Melo P, Kanidou F, Bakirtzis C, Nikolaidis I, Grigoriadou E, Afrantou T, Tatsi T, Matsi S, Grigoriadis N (2019) Induction of apoptosis in CD4(+) T-cells is linked with optimal treatment response in patients with relapsing-remitting multiple sclerosis treated with Glatiramer acetate. J Neurol Sci 401:43–50. https://doi.org/10.1016/j.jns.2019.03.030
Capobianco M, Malucchi S, Ulisciani S, Fava C, Cambrin GR, Avonto L, Saglio G, Bertolotto A (2008) Acute myeloid leukemia induced by mitoxantrone treatment for aggressive multiple sclerosis. Neurol Sci 29(3):185–187. https://doi.org/10.1007/s10072-008-0934-1
Chaudhry V, Cornblat DR, Griffin JW, O’Brien R, Drachman DB (2001) Mycophenolate mofetil: a safe and promising immunosuppressant in neuromuscular diseases. Neurology 56:94–96
Chevret S, Hughes RA, Annane D (2017) Plasma exchange for Guillain-Barre syndrome. Cochrane Database Syst Rev 2:CD001798. https://doi.org/10.1002/14651858.CD001798.pub3
Chitnis T (2007) The role of CD4 T cells in the pathogenesis of multiple sclerosis. Int Rev Neurobiol 79:43–72
Choi K, Hong YH, Ahn SH, Baek SH, Kim JS, Shin JY, Sung JJ (2019) Repeated low-dose rituximab treatment based on the assessment of circulating B cells in patients with refractory myasthenia gravis. Ther Adv Neurol Disord 12:1756286419871187. https://doi.org/10.1177/1756286419871187
Ciafaloni E, Nirjaleshwar K, Nikhar K, Massey JM, Sanders DB (2000) Restrospective analysis of the use of cyclosporine in myasthenia gravis. Neurology 55(3):448–450
Claussen MC, Korn T (2012) Immune mechanisms of new therapeutic strategies in MS: teriflunomide. Clin Immunol 142(1):49–56. https://doi.org/10.1016/j.clim.2011.02.011
Cohen JA, Barkhof F, Comi G, Hartung H-P, Khatri BO, Montalban X, Pelletier J, Capra R, Gallo P, Izquierdo G, Tiel-Wilck K, de Vera A, Jin J, Stites T, Wu S, Aradhye S, Kappos L (2010) Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med 362(5):402–415
Cohen JA, Coles AJ, Arnold DL, Confavreux C, Fox EJ, Hartung H-P, Havrdova E, Selmaj KW, Weiner HL, Fisher E, Brinar VV, Giovannoni G, Stojanovic M, Ertik BI, Lake SL, Margolin DH, Panzara MA, Compston DAS (2012) Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet 380(9856):1819–1828. https://doi.org/10.1016/s0140-6736(12)61769-3
Coles AJ, Twyman CL, Arnold DL, Cohen JA, Confavreux C, Fox EJ, Hartung H-P, Havrdova E, Selmaj KW, Weiner HL, Miller T, Fisher E, Sandbrink R, Lake SL, Margolin DH, Oyuela P, Panzara MA, Compston DAS (2012) Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet 380(9856):1829–1839. https://doi.org/10.1016/s0140-6736(12)61768-1
Comi G, Martinelli V, Rodegher M, Bajenaru O, Carra A, Elovaara I, Fazekas F, Hartung HP, Hillert J, King J, Komoly S, Lubetzki C, Montalban X, Myhr KM, Raavnborg M, Young C, Filippi M (2009) Effect of glatiramer acetate on conversion to clinically definite multiple sclerosis in patients with clinically isolated syndrome (PreCISe study): a randomised, double-blind, placebo-controlled trial. Lancet 274:1503–1511. https://doi.org/10.1016/S0140-
Comi G, Cook S, Giovannoni G, Rieckmann P, Sorensen PS, Vermersch P, Galazka A, Nolting A, Hicking C, Dangond F (2019) Effect of cladribine tablets on lymphocyte reduction and repopulation dynamics in patients with relapsing multiple sclerosis. Mult Scler Relat Disord 29:168–174. https://doi.org/10.1016/j.msard.2019.01.038
Corcione A, Benvenuto F, Ferretti E, Giunti D, Cappiello V, Cazzanti F, Risso M, Gualandi F, Mancardi GL, Pistoia V, Uccelli A (2006) Human mesenchymal stem cells modulate B-cell functions. Blood 107(1):367–372
Costanzi C, Matiello M, Lucchinetti CF, Weinshenker BG, Pittock SJ, Mandrekar J, Thapa P, McKeon A (2011) Azathioprine: tolerability, efficacy, and predictors of benefit in neuromyelitis optica. Neurology 77:659–666
Cree BAC, Lamb S, Morgan K, Chen A, Waubant E, Genain C (2005) An open label study of the effects of rituximab in neuromyelitis optica. Neurology 64(7):1270–1272
Dendrou CA, Fugger L, Friese MA (2015) Immunopathology of multiple sclerosis. Nat Rev Immunol 15(9):545–558. https://doi.org/10.1038/nri3871
Dhib-Jalbut S, Marks S (2010) Interferon-beta mechanisms of action in multiple sclerosis. Neurology 74:S17–S24
Di Ianni M, Del Papa B, De Ioanni M, Moretti L, Bonifacio E, Cecchini D, Sportoletti P, Falzetti F, Tabilio A (2008) Mesenchymal cells recruit and regulate T regulatory cells. Exp Hematol 36(3):309–318. https://doi.org/10.1016/j.exphem.2007.11.007
Dutta R, Trapp BD (2011) Mechanisms of neuronal dysfunction and degeneration in multiple sclerosis. Prog Neurobiol 93(1):1–12. https://doi.org/10.1016/j.pneurobio.2010.09.005
Ersoy E, Kus CNS, Sener U, Coker I, Zorlu Y (2005) The effects of interferong-beta on interleukin-10 in multiple sclerosis patients. Eur J Neurol 12:208–211
Gillard GO, Collette B, Anderson J, Chao J, Scannevin RH, Huss DJ, Fontenot JD (2015) DMF, but not other fumarates, inhibits NF-kappaB activity in vitro in an Nrf2-independent manner. J Neuroimmunol 283:74–85. https://doi.org/10.1016/j.jneuroim.2015.04.006
Gotterer L, Li Y (2016) Maintenance immunosuppression in myasthenia gravis. J Neurol Sci 369:294–302. https://doi.org/10.1016/j.jns.2016.08.057
Greco R, Bondanza A, Vago L, Moiola L, Rossi P, Furlan R, Martino G, Radaelli M, Martinelli V, Carbone MR, Stanghellini MTL, Assanelli M, Bernardi M, Corti C, Peccatori J, Bonini C, Vezzulli P, Falini A, Ciceri F, Comi G (2014) Allogeneic hematopoietic stem cell transplantation for neuromyelitis optica. Ann Neurol 75:447–453. https://doi.org/10.1002/ana
Gross JA, Dillon SR, Mudri S, Johnston J, Littau A, Roque R, Rixon M, Schou O, Foley KP, Haugen H, McMillen S, Waggie K, Schreckhise RW, Shoemaker K, Vu T, Moore M, Grossman A, Clegg CH (2001) TACI-Ig neutralizes molecules critical for B cell development and autoimmune disease: impaired B cell maturation in mice lacking BLyS. Immunity 15:289–302
Group TG-BSS (1985) Plasmaphresis and acute Guillain-Barre syndrome. Neurology 35(8):1096–1104
Harris VK, Stark J, Vyshkina T, Blackshear L, Joo G, Stefanova V, Sara G, Sadiq SA (2018) Phase I trial of intrathecal mesenchymal stem cell-derived neural progenitors in progressive multiple sclerosis. EBioMedicine 29:23–30. https://doi.org/10.1016/j.ebiom.2018.02.002
Hartung H-P, Gonsette R, Konig N, Kwiecinski H, Guseo A, Morrissey SP, Krapf H, Zwingers T (2002) Mitoxantrone in progressive multiple sclerosis: a placebo-controlled, double-blind, randomised, multicentre trial. Lancet 360(9350):2018–2025. https://doi.org/10.1016/s0140-6736(02)12023-x
Hartung HP, Graf J, Kremer D (2019) Long-term follow-up of multiple sclerosis studies and outcomes from early treatment of clinically isolated syndrome in the BENEFIT 11 study. J Neurol. https://doi.org/10.1007/s00415-018-09169-w
Hauser SL, Oksenberg JR (2006) The neurobiology of multiple sclerosis: genes, inflammation, and neurodegeneration. Neuron 52(1):61–76. https://doi.org/10.1016/j.neuron.2006.09.011
Hauser RA, Malek AR, Rosen R (1998) Successful treatment of a patient with severe refractory myasthenia gravis using mycophenolate mofetil. Neurology 51:912–913
Hemmer B, Archelos JJ, Hartung HP (2002) New concepts in the immunopathogenesis of multiple sclerosis. Nat Rev Neurosci 3(4):291–301. https://doi.org/10.1038/nrn784
Hojati Z, Kay M, Dehghanian F (2016) Mechanism of action of interferon beta in treatment of multiple sclerosis. In: Multiple sclerosis. Academic Press, pp 365–392. https://doi.org/10.1016/b978-0-12-800763-1.00015-4
Howard JF, Utsugisawa K, Benatar M, Murai H, Barohn RJ, Illa I, Jacob S, Vissing J, Burns TM, Kissel JT, Muppidi S, Nowak RJ, O’Brien F, Wang JJ, Mantegazza R (2017) Safety and efficacy of eculizumab in anti-acetylcholine receptor antibody-positive refractory generalised myasthenia gravis (REGAIN): a phase 3, randomised, double-blind, placebo-controlled, multicentre study. Lancet Neurol 16(12):976–986
Hughes RAC, Cornblath DR (2005) Guillain-Barré syndrome. Lancet 366(9497):1653–1666. https://doi.org/10.1016/s0140-6736(05)67665-9
Hughes RA, Swan AV, van Doorn PA (2014) Intravenous immunoglobulin for Guillain-Barre syndrome. Cochrane Database Syst Rev 9:CD002063. https://doi.org/10.1002/14651858.CD002063.pub6
Hughes RA, Brassington R, Gunn AA, van Doorn PA (2016) Corticosteroids for Guillain-Barre syndrome. Cochrane Database Syst Rev 10:CD001446. https://doi.org/10.1002/14651858.CD001446.pub5
Jacob A, Matiello M, Weinshenker BG, Wingerchuk DM, Lucchinetti C, Shuster E, Carter J, Keegan BM, Kaantarci OH, Pittock SJ (2009) Treatment of neuromyelitis optica with mycophenolate mofetil - retrospective analysis of 24 patients. Arch Neurol 66(9):1128–1133
Johnson K, Brooks BR, Cohen JA, Ford CC, Goldstein J, Lisak RP, Myers LW, Panitch HS, Rose JW, Schiffer RB (1995) Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. The Copolymer 1 Multiple Sclerosis Study Group. Neurology 45(7):1268–1276
Kappos L, Hartung H-P, Freedman MS, Boyko A, Radü EW, Mikol DD, Lamarine M, Hyvert Y, Freudensprung U, Plitz T, van Beek J (2014) Atacicept in multiple sclerosis (ATAMS): a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Neurol 13(4):353–363. https://doi.org/10.1016/s1474-4422(14)70028-6
Karampera M, Glennie S, Dyson J, Scott D, Laylor R, Simpson E, Dazzi F (2003) Bone marrow mesencyhmal stem cells inhibit the response of naive and memory antigen-specific T cells to their cognate peptide. Blood 101:3722–3729. https://doi.org/10.1182/blood-2002-07-
Karussis D, Petrou P (2018) Immune reconstitution therapy (IRT) in multiple sclerosis: the rationale. Immunol Res 66(6):642–648. https://doi.org/10.1007/s12026-018-9032-5
Karussis DM, Slavin S, Lehmann D, Mizrachi-Koll R, Abramsky O, Ben-Nun A (1992) Prevention of experimental autoimmune encephalomyelitis and induction of tolerance with acute immunosuppression followed by syngeneic bone marrow transplantation? J Immunol 148:1693–1698
Karussis DM, Vourka-Karussis U, Lehmann D, Ovadia H, Mizrachi-Koll R, Ben-Nun A, Abramsky O, Slavin S (1993) Prevention and reversal of adoptively transferred, chronic relapsing experimental autoimmune encephalomyelitis with a single high dose cytoreductive treatment followed by syngeneic bone marrow transplantation. J Clin Invest 92(2):765–772. https://doi.org/10.1172/JCI116648
Kassis I, Grigoriadis N, Gowda-Kurkalli B, Mizrachi-Kol R, Ben-Hur T, Slavin S, Abramsky O, Karussis D (2008) Neuroprotection and immunomodulation with mesenchymal stem cells in chronic experimental autoimmune encephalomyelitis. Arch Neurol 65(6):753–761
Kim SH, Kim W, Li XF, Jung IJ, Kim HJ (2011a) Repeated treatment with rituximab based on the assessment of peripheral circulating memory B cells in patients with relapsing neuromyelitis optica over 2 years. Arch Neurol 68(11):1412–1420. https://doi.org/10.1001/archneurol.2011.154
Kim SH, Kim W, Park MS, Sohn EH, Li XF, Kim HJ (2011b) Efficacy and safety of mitoxantrone in patients with highly relapsing neuromyelitis optica. Arch Neurol 68(4):473–479. https://doi.org/10.1001/archneurol.2010.322
Kim SH, Huh SY, Lee SJ, Joung A, Kim HJ (2013) A 5-year follow-up of rituximab treatment in patients with neuromyelitis optica spectrum disorder. JAMA Neurol 70(9):1110–1117. https://doi.org/10.1001/jamaneurol.2013.3071
Kim SH, Kim Y, Kim G, Park NY, Jang HM, Shin HJ, Hyun JW, Kim HJ (2019) Less frequent rituximab retreatment maintains remission of neuromyelitis optica spectrum disorder, following long-term rituximab treatment. J Neurol Neurosurg Psychiatry 90(4):486–487. https://doi.org/10.1136/jnnp-2018-318465
Kleiter I, Gold R (2016) Present and future therapies in neuromyelitis optica spectrum disorders. Neurotherapeutics 13(1):70–83. https://doi.org/10.1007/s13311-015-0400-8
Kleiter I, Hellwig K, Berthele A, Kumpfel T, Linker RA, Hartung H-P, Paul F, Aktas O (2012) Failure of natalizumab to prevent relapses in neuromyelitis optica. Arch Neurol 69(2):239–245
Kleiter I, Gahlen A, Borisow N, Fischer K, Wernecke KD, Hellwig K, Pache F, Ruprecht K, Havla J, Kumpfel T, Aktas O, Hartung HP, Ringelstein M, Geis C, Kleinschnitz C, Berthele A, Hemmer B, Angstwurm K, Stellmann JP, Schuster S, Stangel M, Lauda F, Tumani H, Mayer C, Krumbholz M, Zeltner L, Ziemann U, Linker R, Schwab M, Marziniak M, Then Bergh F, Hofstadt-van Oy U, Neuhaus O, Zettl UK, Faiss J, Wildemann B, Paul F, Jarius S, Trebst C, Nemos (2018) Apheresis therapies for NMOSD attacks: a retrospective study of 207 therapeutic interventions. Neurol Neuroimmunol Neuroinflamm 5(6):e504. https://doi.org/10.1212/NXI.0000000000000504
Korn T, Mitsdoerffer M, Croxford AL, Awasthi A, Dardalhon VA, Galileos G, Vollmar P, Stritesky GL, Kaplan MH, Waisman A, Kuchroo VK, Oukka M (2008) IL-6 controls Th17 immunity in vivo by inhibitin the conversion of conventional T cells into Foxp3+ regulatory T cells. PNAS 105(47):18460–18465
Kotter MR, Li WW, Zhao C, Franklin RJ (2006) Myelin impairs CNS remyelination by inhibiting oligodendrocyte precursor cell differentiation. J Neurosci 26(1):328–332. https://doi.org/10.1523/JNEUROSCI.2615-05.2006
Krumbholz M, Theil D, Derfuss T, Rosenwald A, Schrader F, Monoranu CM, Kalled SL, Hess DM, Serafini B, Aloisi F, Wekerle H, Hohlfeld R, Meinl E (2005) BAFF is produced by astrocytes and up-regulated in multiple sclerosis lesions and primary central nervous system lymphoma. J Exp Med 201(2):195–200. https://doi.org/10.1084/jem.20041674
Le Page E, Veillard D, Laplaud DA, Hamonic S, Wardi R, Lebrun C, Zagnoli F, Wiertlewski S, Deburghgraeve V, Coustans M, Edan G (2015) Oral versus intravenous high-dose methylprednisolone for treatment of relapses in patients with multiple sclerosis (COPOUSEP): a randomised, controlled, double-blind, non-inferiority trial. Lancet 386(9997):974–981. https://doi.org/10.1016/s0140-6736(15)61137-0
Leist TP, Weissert R (2011) Cladribine: mode of action and implications for treatment of multiple sclerosis. Clin Neuropharmacol 34(1):28–35. https://doi.org/10.1097/WNF.0b013e318204cd90
Liang C, Tian D, Ren X, Ding S, Jia M, Xin M, Thareja S (2018) The development of Bruton’s tyrosine kinase (BTK) inhibitors from 2012 to 2017: a mini-review. Eur J Med Chem 151:315–326. https://doi.org/10.1016/j.ejmech.2018.03.062
Ma Q (2013) Role of Nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol 53(1):401–426. https://doi.org/10.1146/annurev-pharmtox-011112-140320
Mandler RN, Ahmed W, Dencof JE (1998) Devic’s neuromyelitis optica: a prospective study of seven patients treated with prednisone and azathioprine. Neurology 51:1219–1220
Mealy MA, Wingerchuk DM, Palace J, Greenberg BM, Levy M (2014) Comparison of relapse and treatment failure rates among patients with neuromyelitis optica: multicenter study of treatment efficacy. JAMA Neurol 71(3):324–330. https://doi.org/10.1001/jamaneurol.2013.5699
Min JH, Kim BJ, Lee KH (2012) Development of extensive brain lesions following fingolimod (FTY720) treatment in a patient with neuromyelitis optica spectrum disorder. Mult Scler 18:113–115
Minami N, Fujiki N, Doi S, Shima K, Niino M, Kikuchi S, Sasaki H (2011) Five-year follow-up with low-dose tacrolimus in patients with myasthenia gravis. J Neurol Sci 300(1–2):59–62. https://doi.org/10.1016/j.jns.2010.09.033
Mohr DC, Boudewyn AC, Likosky W, Levine E, Goodkin DE (2001) Injectible medication for the treatment of multiple sclerosis: the influence of self-efficacy expectations and injection anxiety on adherence and ability to self-inject. Ann Behav Med 23(2):125–132
Montalban X, Hauser SL, Kappos L, Arnold DL, Bar-Or A, Comi G, de Seze J, Giovannoni G, Hartung HP, Hemmer B, Lublin F, Rammohan KW, Selmaj K, Traboulsee A, Sauter A, Masterman D, Fontoura P, Belachew S, Garren H, Mairon N, Chin P, Wolinsky JS, Investigators OC (2017) Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med 376(3):209–220. https://doi.org/10.1056/NEJMoa1606468
Montalban X, Arnold DL, Weber MS, Staikov I, Piasecka-Stryczynska K, Willmer J, Martin EC, Dangond F, Syed S, Wolinsky JS, Evobrutinib Phase 2 Study Group (2019) Placebo-controlled trial of an oral BTK inhibitor in multiple sclerosis. N Engl J Med 380(25):2406–2417. https://doi.org/10.1056/NEJMoa1901981
Montes Diaz G, Fraussen J, Van Wijmeersch B, Hupperts R, Somers V (2018) Dimethyl fumarate induces a persistent change in the composition of the innate and adaptive immune system in multiple sclerosis patients. Sci Rep 8(1):8194. https://doi.org/10.1038/s41598-018-26519-w
Mulero P, Midaglia L, Montalban X (2018) Ocrelizumab: a new milestone in multiple sclerosis therapy. Ther Adv Neurol Disord 11:1756286418773025. https://doi.org/10.1177/1756286418773025
Muraro PA, Pasquini M, Atkins HL, Bowen JD, Farge D, Fassas A, Freedman MS, Georges GE, Gualandi F, Hamerschlak N, Havrdova E, Kimiskidis VK, Kozak T, Mancardi GL, Massacesi L, Moraes DA, Nash RA, Pavletic S, Ouyang J, Rovira M, Saiz A, Simoes B, Trneny M, Zhu L, Badoglio M, Zhong X, Sormani MP, Saccardi R, Multiple Sclerosis-Autologous Hematopoietic Stem Cell Transplantation Long-term Outcomes Study Group (2017) Long-term outcomes after autologous hematopoietic stem cell transplantation for multiple sclerosis. JAMA Neurol 74(4):459–469. https://doi.org/10.1001/jamaneurol.2016.5867
Nagaishi A, Yukitake M, Kuroda Y (2008) Long-term treatment of steroid-dependent myasthenia gravis patients with low-dose tacrolimus. Intern Med 47(8):731–736. https://doi.org/10.2169/internalmedicine.47.0513
O’Connor P, Wolinsky JS, Confavreaux C, Comi G, Kappos L, Olsson TP, Benzerdjeb H, Truffinet P, Wang L, Miller A, Freedman MS (2011) Randomized trial of oral teriflunomid for relapsing multiple sclerosis. N Engl J Med 365:1203–1303
O’Connor P, Comi G, Freedman MS, Miller AE, Kappos L, Bouchard J-P, Lebrun-Frenay C, Mares J, Benamor J, Thangavelu K, Liang J, Truffinet P, Lawson VJ, Wolinsky JS (2016) Long-term safety and efficacy of teriflunomide: nine-year follow-up of the randomized TEMSO study. Neurology 86:920–930
Palace J, Newsom-Davis J, Lecky B (1998) A randomized double-blind trial of prednisolone alone or with azathioprine in myasthenia gravis. Neurology 50(6):1778–1783
Papadopoulos MC, Verkman AS (2012) Aquaporin 4 and neuromyelitis optica. Lancet Neurol 11(6):535–544. https://doi.org/10.1016/s1474-4422(12)70133-3
Patterson SL, Goglin SE (2017) Neuromyelitis Optica. Rheum Dis Clin N Am 43(4):579–591. https://doi.org/10.1016/j.rdc.2017.06.007
Paty DW, Li DKB (1993) Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. Neurology 43(4):662
Polman CH, O’Connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH, Phillips JT, Lublin FD, Giovannoni G, Wajgt A, Toal M, Lynn F, Panzara MA, Sandrock AW (2006) A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med 354:899–910
Ragheb S, Li Y, Simon KM, VanHaerents S, Galimberti D, De Riz M, Fenoglio C, Scarpini E, Lisak R (2011) Multiple sclerosis: BAFF and CXCL13 in cerebrospinal fluid. Mult Scler J 17(7):819–829
Raivich G, Banati R (2004) Brain microglia and blood-derived macrophages: molecular profiles and functional roles in multiple sclerosis and animal models of autoimmune demyelinating disease. Brain Res Brain Res Rev 46(3):261–281. https://doi.org/10.1016/j.brainresrev.2004.06.006
Ransahoff RM (2007) Natalizumab for multiple sclerosis. N Engl J Med 356:2262–2269
Ransohoff RM, Hafler DA, Lucchinetti CF (2015) Multiple sclerosis-a quiet revolution. Nat Rev Neurol 11(3):134–142. https://doi.org/10.1038/nrneurol.2015.14
Reich DS, Lucchinetti CF, Calabresi PA (2018) Multiple sclerosis. N Engl J Med 378(2):169–180. https://doi.org/10.1056/NEJMra1401483
Rudick RA, Stuart WH, Calabresi PA, Confavreaux C, Galetta SL, Radu E-W, Lublin FD, Weinstock-Guttman B, Wynn DR, Lynn F, Panzara MA, Sandrock AW (2006) Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. N Engl J Med 354:911–923
Schwab N, Schneider-Hohendorf T, Posevitz V, Breuer J, Gobel K, Windhagen S, Brochet B, Vermersch P, Lebrun-Frenay C, Posevitz-Fejfar A, Capra R, Imberti L, Straeten V, Haas J, Wildemann B, Havla J, Kumpfel T, Meinl I, Niessen K, Kleinschnitz C, Warnke C, Buck D, Gold R, Kieseier BC, Meuth SG, Foley J, Chan A, Brassat D, Windel H (2013) L-selection is a possible biomarker for PML risk in natalizumab-treated MS patients. Neurology 81:865–871
Schwab N, Schneider-Hohendorf T, Pignolet B, Spadaro M, Gorlich D, Meinl I, Windhagen S, Tackenberg B, Breuer J, Canto E, Kumpfel T, Hohlfeld R, Siffrin V, Luessi F, Posevitz-Fejfar A, Montalban X, Meuth SG, Gold R, Du Pasquier RA, Kleinschnitz C, Jacobi A, Comabella M, Bertolotto A, Brassat D, Wiendl H (2016) PML risk stratification using anti-JCV antibody index and L-selectin. Mult Scler 22(8):1048–1060
Shenkenberg TD, Von Hoff DD (1986) Mitoxantrone: a new anticancer drug with significant clinical activity. Ann Intern Med 105:67–81
Shimizu Y, Yokoyama K, Misu T, Takahashi T, Fujihara K, Kikuchi S, Itoyama Y, Iwata M (2008) Development of extensive brain lesions following interferon beta therapy in relapsing neuromyelitis optica and longitudinally extensive myelitis. J Neurol 255(2):305–307. https://doi.org/10.1007/s00415-007-0730-5
Sospedra M (2018) B cells in multiple sclerosis. Curr Opin Neurol 31(3):256–262. https://doi.org/10.1097/WCO.000000000000563
Spadaro M, Montarolo F, Perga S, Martire S, Brescia F, Malucchi S, Bertolotto A (2017) Biological activity of glatiramer acetate on Treg and anti-inflammatory monocytes persists for more than 10 years in responder multiple sclerosis patients. Clin Immunol 181:83–88. https://doi.org/10.1016/j.clim.2017.06.006
Tandan R, Hehir MK 2nd, Waheed W, Howard DB (2017) Rituximab treatment of myasthenia gravis: a systematic review. Muscle Nerve 56(2):185–196. https://doi.org/10.1002/mus.25597
Teitelbaum D, Meshhorer A, Hirshfeld T, Arnon R, Sela M (1971) Suppression of experimental allergic encephalomyelitis by a synthetic polypeptide. Eur J Immunol 1:242–248
Thompson AJ, Baranzini SE, Geurts J, Hemmer B, Ciccarelli O (2018) Multiple sclerosis. Lancet 391(10130):1622–1636. https://doi.org/10.1016/s0140-6736(18)30481-1
Tolf A, Fagius J, Carlson K, Akerfeldt T, Granberg T, Larsson EM, Burman J (2019) Sustained remission in multiple sclerosis after hematopoietic stem cell transplantation. Acta Neurol Scand 140(5):320–327. https://doi.org/10.1111/ane.13147
Topakian R, Zimprich F, Iglseder S, Embacher N, Guger M, Stieglbauer K, Langenscheidt D, Rath J, Quasthoff S, Simschitz P, Wanschitz J, Windisch D, Muller P, Oel D, Schustereder G, Einsiedler S, Eggers C, Loscher W (2019) High efficacy of rituximab for myasthenia gravis: a comprehensive nationwide study in Austria. J Neurol 266(3):699–706
Trapp BD, Nave KA (2008) Multiple sclerosis: an immune or neurodegenerative disorder? Annu Rev Neurosci 31:247–269. https://doi.org/10.1146/annurev.neuro.30.051606.094313
Tuohy O, Costelloe L, Hill-Cawthorne G, Bjornson I, Harding K, Robertson N, May K, Button T, Azzopardi L, Kousin-Ezewu O, Fahey MT, Jones J, Compston DA, Coles A (2015) Alemtuzumab treatment of multiple sclerosis: long-term safety and efficacy. J Neurol Neurosurg Psychiatry 86(2):208–215. https://doi.org/10.1136/jnnp-2014-307721
Uccelli A, Laroni A, Brundin L, Clanet M, Fernandez O, Nabavi SM, Muraro PA, Oliveri RS, Radue EW, Sellner J, Soelberg Sorensen P, Sormani MP, Wuerfel JT, Battaglia MA, Freedman MS, MESEMS Study Group (2019) MEsenchymal StEm cells for Multiple Sclerosis (MESEMS): a randomized, double blind, cross-over phase I/II clinical trial with autologous mesenchymal stem cells for the therapy of multiple sclerosis. Trials 20(1):263. https://doi.org/10.1186/s13063-019-3346-z
Van der Meche FGA, Schmitz PIM, The Dutch Guillain-Barre Study Group (1992) A randomized trial comparing intravenous immune globulin and plasma exchange in Guillain-Barre syndrome. N Engl J Med 326:1123–1129
Wingerchuk DMW, Weinshenker BG (2003) Neuromyelitis optica: clinical predictors of a relapsing course and survival. Neurology 60:848–853
Wynn D, Kaufman M, Montalban X, Vollmer T, Simon J, Elkins J, O’Neill G, Neyer L, Sheridan J, Wang C, Fong A, Rose JW (2010) Daclizumab in active relapsing multiple sclerosis (CHOICE study): a phase 2, randomised, double-blind, placebo-controlled, add-on trial with interferon beta. Lancet Neurol 9:381–390. https://doi.org/10.1016/S1474-
Yamasaki R, Lu H, Butovsky O, Ohno N, Rietsch AM, Cialic R, Wu PM, Doykan CE, Lin J, Cotleur AC, Kidd G, Zorlu MM, Sun N, Hu W, Liu L, Lee J-C, Taylor SE, Uehlein L, Dixon D, Gu J, Floruta CM, Zhu M, Charo IF, Weiner HL, Ransohoff RM (2014) Differential roles of microglia and monocytes in the inflamed central nervous system. J Exp Med 211(8):1533–1549. https://doi.org/10.1084/jem.20132477
Yamout B, Hourani R, Salti H, Barada W, El-Hajj T, Al-Kutoubi A, Herlopian A, Baz EK, Mahfouz R, Khalil-Hamdan R, Kreidieh NM, El-Sabban M, Bazarbachi A (2010) Bone marrow mesenchymal stem cell transplantation in patients with multiple sclerosis: a pilot study. J Neuroimmunol 227(1–2):185–189. https://doi.org/10.1016/j.jneuroim.2010.07.013
Zappia E, Casazza S, Pedemonte E, Benvenuto F, Bonanni I, Gerdoni E, Giunti D, Ceravolo A, Cazzanti F, Frassoni F, Mancardi G, Uccelli A (2005) Mesenchymal stem cells ameliorate experimental autoimmune encephalomyelitis inducing T-cell anergy. Blood 106(5):1755–1761. https://doi.org/10.1182/blood-2005-04-1496
Zhang Z, Zhang ZY, Schittenhelm J, Wu Y, Meyermann R, Schluesener HJ (2011) Parenchymal accumulation of CD163+ macrophages/microglia in multiple sclerosis brains. J Neuroimmunol 237(1–2):73–79. https://doi.org/10.1016/j.jneuroim.2011.06.006
Zhang Z, Zhang D, Liu Y, Yang D, Ran F, Wang ML, Zhao G (2018) Targeting Bruton's tyrosine kinase for the treatment of B cell associated malignancies and autoimmune diseases: preclinical and clinical developments of small molecule inhibitors. Arch Pharm (Weinheim) 351(7):e1700369. https://doi.org/10.1002/ardp.201700369
Acknowledgements
We thank the members of the Tsirka lab for their support and helpful discussions. This work was supported by NIH IRACDA K12GM102779.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Thompson, K.K., Tsirka, S.E. (2021). Immunosuppression in Multiple Sclerosis and Other Neurologic Disorders. In: Eisen, H.J. (eds) Pharmacology of Immunosuppression. Handbook of Experimental Pharmacology, vol 272. Springer, Cham. https://doi.org/10.1007/164_2021_545
Download citation
DOI: https://doi.org/10.1007/164_2021_545
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-05117-3
Online ISBN: 978-3-031-05118-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)