
Abstract
Innate immunity exhibits memory characteristics, reflected not only in selective recognition of external microbial or internal damage signals, but more importantly in history and signal-strength dependent reprogramming of innate leukocytes characterized by priming, tolerance, and exhaustion. Key innate immune cells such as monocytes and neutrophils can finely discern and attune to the duration and intensity of external signals through rewiring of internal signaling circuitries, giving rise to a vast array of discreet memory phenotypes critically relevant to managing tissue homeostasis as well as diverse repertoires of inflammatory conditions. This review will highlight recent advances in this rapidly expanding field of innate immune programming and memory, as well as its translational implication in the pathophysiology of selected inflammatory diseases.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Exhaustion
- Inflammatory diseases
- Innate immunity
- Innate memory
- Memory dynamics
- Priming
- Signal strength
- Tolerance
1 Introduction
The establishment of “memory” is the cardinal and classical features of adaptive immunity and has served as the guiding principle of empirical vaccine generation for millennium. Adaptive immunity develops lasting memory responses toward highly specific antigens through somatic recombination-mediated generation of T cell receptors (TCR) and/or B cell receptors (BCR), followed by clonal expansion via interaction with selective antigen presenting cells. In contrast, innate immune cells can only respond to general molecular patterns associated with pathogens through innate receptors (Kawai and Akira 2007). Given limited repertoire of innate receptors, innate immune cells were not historically considered to be memory generating entities. However, emerging data from the last decade reveal fascinating, complex, and dynamic “memory”-like behaviors of innate immune cells that transcend beyond the classical adaptive immune memory phenotypes. The distinct features of innate memory are reflected in signal-strength and history-dependent behaviors such as priming, tolerance, and exhaustion (Li et al. 2020). The generation of innate memory may have profound consequence related to pathophysiology of both acute and chronic inflammatory diseases (Morris et al. 2014).
2 Mechanisms for the Generation of Innate Immune Memory
In the classical sense of immune memory, adaptive immune cells such as T cells and B cells gain the capability to uniquely recognize and memorize highly distinct antigens through somatic VDJ recombination. In sharp contrast, innate immune cells do not have the machinery for VDJ recombination and thus rely upon limited innately encoded receptors to recognize general molecular patterns (e.g., PAMPs – pathogen-associated molecular patterns; DAMPs – damage associated molecular patterns). Despite its limited specificity, innate immune cells can differentiate the signal strength and history of challenges, exhibiting “memory-like” behavior of priming, tolerance, and exhaustion (Geng et al. 2016; Yuan et al. 2016a; Xiong and Medvedev 2011; Foster et al. 2007; Lin et al. 2020). The establishment of such memory-like behavior is clearly distinct from the acquisition of adaptive memory and does not require genetic recombination. Instead, closely intertwined intra-cellular circuitries involving redox signaling, sub-cellular trafficking, metabolic and epigenetic processes are likely involved to establish transient memory states with limited stability and plasticity (Yuan et al. 2016b; Baker et al. 2014, 2015; Maitra et al. 2012; Chan et al. 2005; Netea et al. 2016) (Table 1).
A cardinal example of innate memory can be seen with monocyte/macrophage responses to rising dosages of bacterial endotoxin (Yuan et al. 2016b; Lu et al. 2015). While a prolonged challenge with higher dosages of lipopolysaccharide (LPS) can lead to reduced expression of pro-inflammatory cytokines, commonly known as endotoxin tolerance (Morris et al. 2014), prolonged stimulation with a subclinical super-low dose LPS can polarize monocyte/macrophage into a “primed” low-grade inflammatory state with sustained expression of inflammatory mediators (Yuan et al. 2016a, b). The mechanisms of endotoxin tolerance likely involve the activation and induction of molecular suppressors at multiple levels such as cytoplasmic signaling suppressors interleukin-1R-associated-kinase (IRAK)-M, and phosphatidylinositol-3-kinase and protein kinase B (PI3K/AKT) (Xiong and Medvedev 2011; Piao et al. 2009), as well as nuclear transcriptional suppressor RelB (Maitra et al. 2012; Chan et al. 2005). On the other hand, the generation of primed low-grade inflammatory monocyte/macrophage requires the clearance of suppressors such as IRAK-M and PI3K/AKT (Geng et al. 2016; Maitra et al. 2012). At the sub-cellular level, subclinical super-low dose LPS preferentially disrupts the homeostatic processes of autophagic flux as well as pexophagy, leading to the accumulation of reactive oxygen species involved in the establishment of low-grade inflammation (Yuan et al. 2016a; Geng et al. 2019). Innate leukocytes may sense the signal strength and duration of LPS via distinct usage and assembly of intra-cellular adaptor molecules such as myeloid differentiating factor 88 (MyD88) and TRIF-related adaptor molecule (TRAM), with TRAM preferentially directing the cellular response to sustained stimulation of super-low dose LPS (Yuan et al. 2016b; Rahtes and Li 2020). On the other hand, MyD88 is preferentially involved in response to higher dose LPS during both the acute response phase and the compensatory phase of tolerance (Cheng et al. 2015; Laird et al. 2009). The intra-cellular processes responsible for priming and tolerance may likely compete with each other forming multi-tiered competitive circuitries, assisting the decision-making processes of innate leukocytes in adopting dynamic activation behaviors (Morris et al. 2014; Fu et al. 2012; Kadelka et al. 2019) (Fig. 1). The generation of mutually competitive circuitries is also a fundamental principle for the clear differentiation and activation of other immune cells such as T helper cells (Hong et al. 2011, 2012).

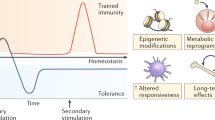
Illustration of innate memory dynamics based on signal strength and duration. Innate immune leukocytes such as monocytes and macrophages can finely sense the strength and duration of external danger signals (e.g., lipopolysaccharide, LPS) and undergo distinct adaptations to generate dynamic memory states. In the case of LPS, a prolonged challenge with subclinical super-low dose LPS (<1 ng/mL) will induce a sustained low-grade inflammatory states due to the positive-feedback signals involving mutually activating TRAM adaptor, SRC kinases, IRF1/5/7, and STAT1. In contrast, while higher dose LPS acutely induces a transient and robust inflammatory response through the activation of NFkB, prolonged stimulation with higher LPS signals will trigger the expression of inhibitory kBs (IkBs) and RelB, leading to a tolerant state with reduced expression of inflammatory mediators such as CD86, CD40, and CCR5. Tolerant leukocytes still maintain a skewed expression of profile of selected immune suppression genes such as PD-L1, and eventually adopt an exhausted state characterized by pathogenic inflammation and immune suppression
Sustained challenges with higher dose endotoxin lead to not only endotoxin tolerance, but also an exhausted state characterized by pathogenic inflammation and immunosuppression often seen during the progression of sepsis (Efron et al. 2018; Horiguchi et al. 2018). “Endotoxin tolerant” cells are not inert and can still robustly respond to endotoxin stimulation, with a significantly altered landscape of gene expression potentially contributing to pathogenic inflammation and immune exhaustion (Foster et al. 2007; Lu et al. 2015). For example, monocyte/macrophage with prolonged LPS stimulation exhibits robust induction of iNOS and PD-L1 (Foster et al. 2007; Lu et al. 2015). Persistent iNOS expression may contribute to pathogenic inflammation, and PD-L1 is a major contributor mediating immune suppression. Recently, we demonstrate that endotoxin exhaustion is not limited to monocyte/macrophage and can also be seen in neutrophils with prolonged challenge of higher dose LPS (Lin et al. 2020). Exhausted neutrophils with prolonged LPS treatment manifest enhanced expression of pathogenic inflammatory mediators such as LTB4 and ICAM1, contributing to altered migratory and swarming behaviors reminiscing septic neutrophils (Lin et al. 2020). Exhausted neutrophils similarly express elevated PD-L1, potentially contributing to immune suppression (Lin et al. 2020).
3 Innate Immune Memory During the Pathogenesis of Acute and Chronic Diseases
3.1 Low-Grade Inflammatory Memory Monocyte in Atherosclerosis
Atherosclerosis and related cardiovascular complications are among the leading causes of morbidity and mortality in the world (Libby et al. 2019). Previously considered as a lipid storage disease, atherosclerosis is nowadays well recognized as a chronic low-grade inflammatory disease that occurs within the arterial wall (Back et al. 2019). The programming of low-grade inflammatory monocytes is crucially involved in the pathogenesis of atherosclerosis. Non-resolving low-grade inflammatory monocytes and monocyte-derived macrophages are the key mediators for the formation and progression of atherosclerotic plaques (Jongstra-Bilen et al. 2006; Libby and Hansson 2015). Monocytes can be primed by risk factors present in the circulation and in the vessel wall, such as pathogen-associated molecular patterns, oxidized lipoproteins, shear stress, and oxidative stress. Excessive inflammatory signals tend to trigger compensatory anti-inflammatory tolerance and therefore the expression of pro-inflammatory mediators in monocytes is transient and subsequently suppressed due to the induction of homeostatic negative regulators (Nathan and Ding 2010; Biswas and Lopez-Collazo 2009; Adib-Conquy and Cavaillon 2009). In contrast, under non-resolving low-grade inflammatory conditions, monocytes may fail to develop tolerance and are programmed into a sustained inflammatory state that favors the development of atherosclerosis (Baker et al. 2014; Maitra et al. 2012; Deng et al. 2013).
LPS, also known as endotoxin, is the major stimulant to prime monocytes, which are the primary immune cells responding to LPS given their relatively high expression of TLR4. Trace amount of gut microbiota-derived LPS may leak into circulation via increased gut permeability, leading to subclinical endotoxemia (Frazier et al. 2011; Lassenius et al. 2011). According to epidemiological studies endotoxemia levels as low as 50 pg/mL may serve as a strong risk factor for the development of atherosclerosis (Stoll et al. 2004). Indeed, atherosclerosis patients have low but significantly elevated serum LPS level as compared with healthy individuals (79.0 ± 10.7 vs. 43.5 ± 11.9 pg/mL, p < 0.001). This concentration of LPS is sufficient to up-regulate Nox2 expression and elevate oxidative stress in human monocytes (Carnevale et al. 2018). In the murine model of atherosclerosis, ApoE−/− mice fed with high-fat diet exhibit significantly higher level of serum LPS as compared to the counterparts fed with regular diet. Oral administration of Akkermansia muciniphila decreases the circulating LPS level, alleviates atherosclerosis progression, as well as reduces monocyte/macrophage accumulation in the plaques (Li et al. 2016). These findings indicate that low-grade inflammatory monocytes primed by low-dose LPS are critically involved in the pathogenesis of atherosclerosis.
Chronic injection of subclinical dose LPS to high-fat diet-fed ApoE−/− mice (a murine model of atherosclerosis) significantly exacerbates the pathogenesis of atherosclerosis accompanied by higher levels of circulating Ly6CPositive low-grade inflammatory monocytes as well as increased number of macrophages within the plaque areas. The surface level of inflammatory chemotaxis receptor CCR5 is significantly elevated while the surface expression of SR-B1, a modulator for anti-inflammation and lipid metabolism, is reduced on circulating monocytes from the high-fat diet-fed ApoE−/− mice conditioned with super-low dose LPS. The monocytes that are primed with subclinical dose LPS for a long-term exhibit similar phenotype, as characterized by enhanced levels of CCR5 and reduced levels of SR-B1. Adoptive transfer of these LPS primed monocytes to high-fat diet-fed ApoE−/− mice results in significant elevation of plaque size and lipid deposition, suggesting that these low-grade inflammatory monocytes programmed by subclinical dose LPS can directly contribute to atherosclerosis progression. Mechanistically, super-low dose LPS treatment induces increased level of miR-24, which mediates the suppression of SR-B1, and reduction of IRAK-M, which is a critical negative-feedback regulator. IRAK-M deficiency in turn leads to elevated miR-24 levels, forming a positive-feedback loop sustaining the low-grade inflammatory state conducive to atherosclerosis (Geng et al. 2016). There are two competitive pathways transducing signals following LPS stimulation, namely the MyD88-dependent pathway and the MyD88-independent pathway mediated by TRIF and TRAM (Palsson-McDermott and O’Neill 2004). Intriguingly, the low-grade inflammatory monocyte primed by super-low dose LPS is dependent upon TRAM/TRIF but not MyD88 (Yuan et al. 2016b). By employing a bone marrow transplantation strategy, Lundberg et al. have shown that hematopoietic deficiency of TRAM and TRIF but not MyD88 adaptor-like (MAL) significantly reduces atherosclerosis in Ldlr−/− mice (another murine model of atherosclerosis). TRAM deficiency also leads to down-regulated level of pro-inflammatory mediators, such as TNF-α, IL-6, IL-12, CCL2, CCL5, and CXCL10, in the aorta of atherosclerotic mice (Lundberg et al. 2013). These data suggest that the priming of low-grade inflammatory monocytes by subclinical dose LPS during atherosclerosis is mainly mediated by TRAM, and targeting TRAM may promote effective generation of resolving monocytes for the prevention and treatment of atherosclerosis.
In addition to low-dose LPS, low concentrations of oxidized low-density lipoprotein (oxLDL) can also induce epigenetic reprogramming of monocytes into a pro-inflammatory state. Primary human monocytes trained with low doses of oxLDL (below 10 μg/mL) for 24 h exhibit an enhanced response to secondary stimulation 6 days later by expressing a series of pro-inflammatory mediators, including IL-6, TNFα, IL-8, MCP-1, MMP-2, and MMP-9. These trained monocytes have enhanced capacity to generate foam cells, elevated expressions of scavenger receptors (CD36 and SR-A), and reduced expression of cholesterol efflux transporters (ABCA1 and ABCG1). Therefore, these pro-inflammatory monocytes may contribute to the pathogenesis of atherosclerosis. The oxLDL-induced long-lasting proatherogenic profile can be significantly attenuated if the monocytes are pre-treated with histone methyltransferase inhibitor, suggesting that epigenetic histone modification is crucial for this innate immune memory of monocytes (Bekkering et al. 2014). It has been found that oxLDL treatment can cooperatively boost the activation of macrophages induced by low-dose LPS. Costimulation with oxLDL and low-dose LPS significantly up-regulates the genes transcribed by promoters containing an AP-1 binding site as well as induces the activation of ERK1/2. The combined effects of subclinical endotoxemia and oxLDL result in the establishment of pro-inflammatory state of macrophages and production of a series of inflammatory cytokines within atherosclerotic lesions (Wiesner et al. 2010).
3.2 Exhausted Memory Innate Leukocytes During the Pathogenesis of Sepsis
Sepsis is a systemic inflammatory response to severe infection and injury leading to multi-organ failure and remains one of the primary causes of death in hospitalized patients (Rhee et al. 2019; Perner et al. 2016). In 2017, global incidence of sepsis was around 48.9 million cases and sepsis-related deaths were estimated at 11.0 million cases (Rudd et al. 2020). The new coronavirus (SARS-CoV-2) in the ongoing outbreak and its associated disease COVID-19 pose tremendous threats to public health and drastically affect worldwide economies and societies (Kumar 2021a, b). Particularly, sepsis is the leading cause of death by COVID-19, which has been observed in nearly all deceased patients in numerous cohorts (Lopez-Collazo et al. 2020; Kumar 2020). The immune response of sepsis patients consists of a hyperinflammatory phase featured by “cytokine storm” and an immunosuppressive phase exemplified by immune cell exhaustion and dysfunction (Hotchkiss et al. 2016). Many clinical trials have been conducted to attenuate the hyperinflammatory effects by using anti-cytokine or anti-inflammatory agents, such as anti-IL-1β, anti-TNF-α, anti-LPS, and TLR inhibitors. Unfortunately, none of these approaches produces robust curative outcomes, and in some cases, the survival rate was even reduced (Brady et al. 2020; Abraham et al. 1997; Opal et al. 2013). A hallmark of sepsis is diminished clearance of primary pathogens and increased risk of secondary infection due to pathogenic inflammation and immune suppression (Efron et al. 2018). Over 70% of deaths occur after the first 3 days of sepsis, many of which occur weeks after sepsis onset (Otto et al. 2011). Thus, immunosuppression caused by leukocyte exhaustion has been increasingly recognized as a major factor for sepsis-induced mortality. A recent single cell study revealed that moribund COVID patients tend to have higher numbers of exhausted classical monocytes (Schulte-Schrepping et al. 2020).
T cell exhaustion driven by persistent exposure to infections during sepsis has been well documented in the literature. The exhausted T cells are defined by a progressive loss of T cell effector function, a state of vigilant transcription distinct from functional effector or memory T cells. A typical alteration of exhausted T cells is the overexpression of a series of inhibitory molecules, such as PD-1, CTLA-4, LAG-3, and TIM-3 (Wherry 2011). PD-1 is a critical negative regulator involved in suppressing lymphocyte responses. PD-1/PD-L1 pathway plays an important role in the initiation and promotion of immunosuppression (Liu and Li 2017). Multiple studies using mouse model of cecal ligation and puncture (CLP) have unveiled elevated PD-1 expression on splenic CD4+ and CD8+ T cells. There is a continuously increased PD-1 expression on CD4+ and CD8+ T cells with the progression of sepsis, associated with a drastic reduction of total T cell population. Similarly, sepsis patients also have significantly increased PD-1 expression on T cells in the peripheral blood, spleen as well as injured organs (Patil et al. 2017). These exhausted T cells from sepsis patients fail to efficiently produce inflammatory cytokines and their secretory profiles are potently compromised (O’Sullivan et al. 1995; Wick et al. 2000; Heidecke et al. 1999). The ploy-functionality of CD8+ cells is also significantly impaired in severe sepsis patients, and PD1 expression is inversely correlated with the number of poly-functional CD8+ T cells (Choi et al. 2017). PD-1 is considered as one of the most promising targets for immunomodulatory therapy to resume T cell function. However, anti-PD-1 treatment alone does not yield expected outcomes because multiple negative costimulatory molecules are expressed on the surface of exhausted T cells. For example, a recent study demonstrates that T cells co-expressing LAG3 and PD-1 are more significantly exhausted as compared to LAG3 or PD-1 single positive T cells in patients with acute sepsis. Furthermore, the frequency of co-expressing T cells is positively associated with the mortality and the length of hospital stay (Niu et al. 2019). Thus, therapies targeting these suppressor molecules may maximize the recovery of T cells.
Correspondingly, monocytes in sepsis patients tend to express higher levels of immune receptors including CD63, CD163, CD206, TLR2, and TLR4, presumably rending them with elevated responses to infections (Armstrong et al. 2004; Hirsh et al. 2001). However, studies reveal that monocytes from sepsis patients are less responsive than those from healthy individuals. They cannot efficiently produce TNF-α, IL-1β, and IL-8 when challenged with LPS ex vivo, and the reduced TNF-α production by monocytes is employed as an index to evaluate the immune suppression of patients with sepsis (Ryan et al. 2017). The diminished capacity to produce pro-inflammatory cytokines may be due to the elevated expression of IRAK-M, an inhibitory Toll receptor signaling molecule, in the monocytes from sepsis patients. The patients with higher IRAK-M levels on admission have a higher mortality rate (Wiersinga et al. 2009). Monocytes are specialized antigen presenting cells (APCs) that present surface MHC molecule-bound antigens to activate T cells. The exhaustion of these APCs potentially facilitates the immunosuppression during sepsis. Sepsis induces altered monocyte–T cell interactions because of reduced expressions of co-stimulatory molecules on monocytes. Indeed, monocytes in sepsis patients are found to express much lower levels of CD40, CD80, and CD86 (Sugimoto et al. 2003; Sinistro et al. 2008; Lissauer et al. 2009). On the contrary, PD-L1 surface expression is up-regulated on monocytes from septic mice models as well as sepsis patients, correlated with T cell exhaustion and immunosuppression via PD-1/PD-L1 signaling pathway (Patil et al. 2017). Monocyte PD-L1 expression can be used as an independent predictor of 28-day mortality in patients with septic shock (Shao et al. 2016).
Neutrophils are the most abundant leukocytes in the circulation and play a crucial role in sepsis as the first line of defense in protecting the body from microbial invasion. The interaction between neutrophils and other immune cells is necessary for the resolution of excessive inflammation as well as effective host defense (Serhan and Savill 2005). Exhausted neutrophils with aberrant immune responses to infection have been observed in septic animal models and patients. Excessive bacterial products and pro-inflammation cytokines in sepsis induce the loss of CD62L expression but elevated integrin expression (e.g., CD11b) on the surface of neutrophils (Rosenbloom et al. 1999; Kovach and Standiford 2012). In addition, CXCR2 expression is reduced in the neutrophils of mice and patients with severe sepsis. Prolonged neutrophil stimulation results in up-regulation of iNOS and GRK2, which further promotes CXCR2 internalization (Paula-Neto et al. 2011). Therefore, neutrophils exhibit reduced rolling and migratory capacity and fail to be recruited to the primary infection foci. Instead, these exhausted neutrophils tend to form pathogenic aggregations in the vital organs, which can be mimicked through in vitro examination (Lin et al. 2020). The antimicrobial activities of neutrophils are also compromised during sepsis. Excessive bacterial load activates complement system, and high levels of C5a suppress the phagocytic function and ROS production of neutrophils. Various studies with septic mouse models and sepsis patients have revealed impaired phagocytosis, oxidant production as well as oxidative burst capacity of septic neutrophils (Shen et al. 2017; Bhan et al. 2016). Furthermore, neutrophils from septic mice and patients can induce immunosuppression and T cell apoptosis in a cell-contact dependent manner via the surface expression of PD-L1. Thus, the PD-L1 level on neutrophils, which is positively associated with sepsis severity, may serve as a biomarker for the prognosis of septic patients (Wang et al. 2015). Despite its clinical significance, the mechanisms of neutrophil exhaustion during sepsis are still poorly understood. We have recently reported that neutrophils treated with LPS in vitro for a prolonged period develop a phenotype of elevated ICAM1, CD11b, and PD-L1 expression as well as enhanced swarming and aggregation, which resembles the exhausted neutrophil phenotypes seen in sepsis. Importantly, the exhaustive profiles are significantly alleviated in TRAM deficient neutrophils after prolonged LPS challenge as compared with wild-type neutrophils. TRAM mediated neutrophil exhaustion may be dependent upon Src family kinases (SFK) and STAT1 activation, since SFK inhibitor can effectively block neutrophil exhaustion caused by prolonged LPS treatment. Furthermore, TRAM deficiency is protective against the development of severe systemic inflammation and multi-organ damage in mice (Lin et al. 2020). These data unveil a critical function of TRAM in promoting neutrophil exhaustion.
3.3 Innate Immune Memory During the Pathogenesis of Cancer
The phenotype and functionalities of myeloid cells (e.g., monocytes, macrophages, and neutrophils) are substantially changed by tumor-induced systemic environment and microenvironment, so that these cells usually acquire pro-tumor functions to promote cancer progression. On the other hand, the tumor-associated innate immune cells may provide ideal targets for fighting against cancer.
The number of circulating monocytes significantly increases in both humans and mice bearing tumors. Among several cancer types, patients with high blood monocyte counts have a poorer disease prognosis, and the ratio of lymphocytes to monocytes has become a prognostic factor for lung cancer, colorectal cancer, and ovarian cancer (Kiss et al. 2020; Olingy et al. 2019). The increased monocyte levels may be caused by two reasons: enhanced migration from bone marrow to circulation, and increased myelopoiesis. Patients with pancreatic cancer exhibit elevated circulating monocyte levels associated with decreased monocyte abundance in the bone marrow. CCL2, a critical chemokine for monocyte recruitment, is commonly present at higher levels in serum of both mice and humans with cancer, facilitating the egress of monocytes from the bone marrow (Kishimoto et al. 2019). Cancer is usually accompanied by elevated serum levels of cytokines that are involved in the myeloid cell differentiation and survival, such as macrophage-colony stimulating factor (M-CSF), granulocyte-colony stimulating factor (G-CSF), and granulocyte-macrophage-colony stimulating factor (GM-CSF) (Scholl et al. 1996; Ribechini et al. 2017; Katsumata et al. 1996). Excessive production of these cytokines and tumor-associated low-grade inflammation promote reprogramming of myelopoiesis. As a result, hematopoietic stem cell (HSC), common myeloid progenitor (CMP), and granulocyte-monocyte progenitor (GMP) populations are expanded, while common lymphoid progenitor (CLP) and megakaryocyte-erythrocyte progenitor (MEP) are not significantly altered. For example, increased frequency of HSC and GMP populations is observed in peripheral blood of patients with various types of solid tumors, indicating that tumor-associated environment favors myeloid hematopoiesis and expansion of circulating monocytes (Wu et al. 2014; Manz and Boettcher 2014; Casbon et al. 2015; Strauss et al. 2020). In addition to increased numbers, the phenotype of monocytes is also profoundly influenced by tumors. One of the well-documented features of cancer-educated monocytes is the acquisition of immunosuppressive properties. The monocytes from healthy individuals express high level of HLA-DR (the protein of MHC II) on the surface, while HLA-DR level is significantly down-regulated in the monocytes of cancer patients (Luczynski et al. 2004; Ugurel et al. 2004). The high level of CD14 HLA-DRlow monocytes is correlated with the lower levels of tumor-specific T-cells in the circulation of cancer patients. The patients with lower levels of CD14+ HLA-DRlow monocytes are more responsive to immune checkpoint blockade therapy (Weide et al. 2014; Weber et al. 2016). The surface expression of CD86, a co-stimulatory molecule for T cell activation, is also reduced on cancer-educated monocytes, inhibiting T cell function (Luczynski et al. 2004; Ugurel et al. 2004). Furthermore, monocytes in cancer patients have enhanced expression and activity of arginase-1, which limits the availability of L-arginine to T cells (Trovato et al. 2019; Hoechst et al. 2008). Up-regulation of PD-L1 and GPNMB may also contribute to the immunosuppressive activity of monocytes in cancer (Kobayashi et al. 2019). Intriguingly, monocytes from cancer patients exhibited increased level of phosphorylated STAT3, and STAT3 is potentially activated in the healthy monocytes after co-culture with cancer cells. Inhibition of STAT3 attenuates the immunosuppressive activity of cancer-educated monocytes (Trovato et al. 2019; Poschke et al. 2010). Therefore, STAT3 may be a crucial transcription factor for the reprogramming of monocytes by tumor-specific environment.
Based on the pro-tumor characteristics of monocytes, a series of regimens have been developed to reverse monocyte reprogramming. Specific antibody against CCR2 or small molecules that block CCR2 signaling remarkably restrain the growth and metastasis of tumor cells in mouse models of lung, breast, prostate, pancreatic, and liver cancers (Olingy et al. 2019). A recent clinical study has revealed that oral administration of PF-04136309, a small molecule CCR2 antagonist, effectively reduces circulating monocytes and tumor-associated macrophages (TAMs) in pancreatic cancer (Nywening et al. 2016). Angiotensin II serves as a key regulator of cancer-induced myelopoiesis, and angiotensin-converting enzyme (ACE) inhibitors may suppress the excessive generation of pro-tumor monocytes. Enalapril, an ACE inhibitor, has been shown to reduce monopoiesis and TAMs as well as prolong the survival of mice with lung tumors (Cortez-Retamozo et al. 2013). Neutralization of tumor-derived GM-CSF reduces the emergence of CD11b+ Gr-1+ myeloid cells, leading to elevated anti-tumor activity of T cells and restrained tumor growth (Bayne et al. 2012). Arginase inhibition may also be an alternative approach to mitigate monocyte-mediated arginine depletion and consequent immunosuppression. Accordingly, a small molecule arginase inhibitor has been developed, which can increase plasma and tumor arginine levels, enhance anti-tumor T cell responses, prime immunity toward a pro-inflammatory state, and reduce tumor growth in mouse cancer models (Steggerda et al. 2017). Application of STAT3 inhibitors has also yielded promising anti-tumor outcomes by boosting anti-tumor immunity in mice. TAM receptor tyrosine kinases promote STAT3 phosphorylation, and administration of UNC4241, an inhibitor against TAM receptor tyrosine kinases, significantly alleviates the immunosuppressive activity of monocytes in a mouse model of melanoma (Holtzhausen et al. 2019).
Neutrophil is one of the major components of tumor-infiltrating innate immune cells. Cancer patients with poor prognosis often have an expanded pool of tumor-associated neutrophils (TANs), which exhibit complex and contradictory functions, promoting or limiting tumor growth (Powell and Huttenlocher 2016). The pro-tumor neutrophils produce high levels of VEGF, MMP9, and HGF, which facilitate tumor angiogenesis. MMP9 production and NET formation from neutrophils may provide ideal environment and niche for tumor cell intravasation and metastasis. Similar as cancer-educated monocytes, some TANs also possess immunosuppressive properties and suppress T cell proliferation via deprivation of L-arginine (Granot 2019). Compared with circulating and splenic neutrophils, TANs secrete higher amount of CCL7 (a Tregs chemoattractant) that promotes the recruitment of Tregs to tumor, thereby forming an immunosuppressive microenvironment (Fridlender and Albelda 2012). On the other hand, some neutrophils also exert anti-tumor activities by secreting cytotoxic mediators (ROS) to induce tumor cell apoptosis (Granot 2019). Intriguingly, accumulating data have suggested that the interaction between neutrophils and T cells is indispensable for the proper anti-tumor response of adaptive immunity (Eruslanov et al. 2014; Stoppacciaro et al. 1993). A subset of TANs has been found to exhibit both neutrophil and APC characteristics in the patients with lung cancer, and these cells can boost anti-tumor T cell responses (Singhal et al. 2016). The molecular mechanisms underlying the differential function of neutrophils are not well understood. A recent study has indicated that Tollip, an innate immunity signaling adaptor molecule, may contribute to the neutrophil reprogramming in a mouse model of colorectal cancer. Tollip-deficient neutrophils have STAT5-dependent elevation of CD80 and reduction of PD-L1 as compared to wild-type counterparts. Therefore, Tollip-deficient neutrophils may potently activate T cells to exert anti-tumor activity. Adoptive transfer of Tollip-deficient neutrophils but not Tollip-deficient monocytes promotes tumor immune surveillance and reduces colorectal cancer burden in vivo. Thus, Tollip may serve as a target to modulate the decision-making process of neutrophils for future cancer therapy (Zhang et al. 2019).
In recent years, the concept of trained immunity has drawn increasing attention due to its potential for the treatment of diseases such as cancer. Trained immunity represents an epigenetic and metabolic reprogramming process of innate immune cells to acquire long-term function and memory property (Netea et al. 2017). Instillation of Bacillus Calmette-Guérin (BCG) can prime monocytes from the patients with bladder cancer into a pro-inflammatory and anti-cancer profile, mediated by the autophagy genes ATG2B and ATG5 (Buffen et al. 2014). The patients who exhibit low responsiveness to trained immunity have a higher chance of recurrence and tumor progression (Netea et al. 2016). In LPS primed macrophages, 70% of the inducible genes are the targets of Jumonji domain containing 3 (JMJD3), also known as lysine demethylase 6B (KDM6B) (De Santa et al. 2009). KDM6B can stabilize tumor suppressor gene p53 (Ene et al. 2012), and higher KDM6B expression is a prognostic indicator for better survival in neuroblastoma patients (Yang et al. 2019). Thus, LPS-trained macrophages via KDM6B may have clinical potential for cancer therapy. Fungal-derived polysaccharide β-glucans have been used to treat various cancers for a long time, and they are also potential inducers to promote trained immunity. A series of studies have revealed that β-glucan treatment leads to marked phenotypical and functional alterations of monocytes/macrophages, such as elevated cytokine production, enhanced phagocytic capacity, and increased ROS generation (Netea et al. 2017; Lerias et al. 2019). A recent study has shown that training with β-glucan leads to a transcriptomic and epigenetic rewiring of granulopoiesis in mice with melanoma, which subsequently induces the anti-tumor phenotype of TANs. Importantly, the mice receiving a single injection of β-glucan still exhibit a significant inhibition of tumor growth after 28 days, indicating long-term anti-tumor effects of neutrophil-mediated trained Immunity (Kalafati et al. 2020).
4 Concluding Remarks
Despite these exciting advances, the field of innate immune memory has only been revealed as the very tip of an iceberg closely intertwined with almost all aspects of human pathophysiology. Innate leukocyte may adopt highly diverse disease and context-dependent memory states, requiring single cell approaches to further clarify their unique contribution to distinct pathogenesis of inflammatory diseases. Memory leukocytes may further propagate their phenotypes to neighboring cells, establishing unique memory niche within local environments (Ballesteros et al. 2020). Future efforts are needed to finely map the establishment as well as propagation of innate memory through genetic and chemical approaches in tracking the ontogeny and propagation of memory innate leukocytes, in response to varying degrees of signal strengths and intensities within complex host immune environments. Harnessing the potential of reprograming innate memory would hold enormous promise in the treatment of both acute and chronic human diseases.
Abbreviations
- ABCA1:
-
ATP-binding cassette sub-family A member 1
- ABCG1:
-
ATP-binding cassette sub-family G member 1
- ACE:
-
Angiotensin-converting enzyme
- APC:
-
Antigen presenting cells
- ApOE:
-
Apolipoprotein E
- ATG:
-
Autophagy-related gene
- BCG:
-
Bacillus Calmette-Guérin
- BCR:
-
B cell receptor
- CCL:
-
C-C motif chemokine ligand
- CCR:
-
C-C Chemokine receptor
- CLP:
-
Cecal ligation and puncture
- CMP:
-
Common myeloid progenitor
- CTLA-4:
-
Cytotoxic T-lymphocyte-associated protein 4
- CXCL:
-
C-X-C motif chemokine ligand
- CXCR:
-
C-X-C motif chemokine receptors
- DAMP:
-
Damage-associated molecular pattern
- ERK1/2:
-
Extracellular signal-regulated kinase 1/2
- G-CSF:
-
Granulocyte-colony stimulating factor
- GM-CSF:
-
Granulocyte-macrophage colony stimulating factor
- GMP:
-
Granulocyte-monocyte progenitors
- GPNMB :
-
Glycoprotein-Nmb
- GRK2:
-
G protein-coupled receptor kinases
- HGF:
-
Hepatocyte growth factor
- HSC:
-
Hematopoietic stem cell
- ICAM1:
-
Intercellular adhesion molecule 1
- IL:
-
Interleukin
- iNOS:
-
Inducible nitric oxide synthase
- IRAK-M:
-
Interleukin-1R-associated-kinase- M
- JMJD3:
-
Jumonji domain containing 3
- KDM6B:
-
Lysine demethylase 6B
- LAG-3:
-
Lymphocyte-activating gene
- Ldlr:
-
Low-density lipoprotein receptor
- LPS:
-
Lipopolysaccharide
- LTB4:
-
Leukotriene B4
- MAL:
-
MyD88-adapter-like
- MCP:
-
Monocyte chemoattractant protein
- M-CSF:
-
Macrophage-colony-stimulating factor
- MEP:
-
Megakaryocyte-erythrocyte progenitor
- MMP:
-
Matrix metalloproteinases
- MYD88:
-
Myeloid differentiation factor 88
- NET:
-
Neutrophil extracellular trap
- NOX2:
-
NADPH oxidase 2
- oxLDL:
-
Oxidized low-density lipoprotein
- PAMP:
-
Pathogen associated molecular pattern
- PD-1:
-
Programmed cell death protein-1
- PD-L1:
-
Programmed death-ligand 1
- PI3K/AKT:
-
Phosphatidylinositol-3-kinase and protein kinase B
- SFK:
-
Src family kinases
- SR-A:
-
Scavenger receptor class A
- SR-B1:
-
Scavenger receptor class B type 1
- STAT:
-
Signal transducer and activator of transcription
- TAM:
-
Tumor-associated macrophages
- TAN:
-
Tumor-associated neutrophils
- TCR:
-
T cell receptor
- TIM-3:
-
T-cell immunoglobulin and mucin-domain containing-3
- TLR:
-
Toll-like receptor
- TNF:
-
Tumor necrosis factor
- TRAM:
-
Toll/IL-1R domain-containing adaptor-inducing IFN-β-related adaptor molecule
- TRIF:
-
Toll/IL-1R domain-containing adaptor-inducing IFN-β
- VEGF:
-
Vascular endothelial growth factor
References
Abraham E et al (1997) p55 tumor necrosis factor receptor fusion protein in the treatment of patients with severe sepsis and septic shock. A randomized controlled multicenter trial. Ro 45-2081 study group. JAMA 277:1531–1538
Adib-Conquy M, Cavaillon JM (2009) Compensatory anti-inflammatory response syndrome. Thromb Haemost 101:36–47
Armstrong L, Medford AR, Hunter KJ, Uppington KM, Millar AB (2004) Differential expression of toll-like receptor (TLR)-2 and TLR-4 on monocytes in human sepsis. Clin Exp Immunol 136:312–319
Back M, Yurdagul A Jr, Tabas I, Oorni K, Kovanen PT (2019) Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat Rev Cardiol 16:389–406
Baker B, Maitra U, Geng S, Li L (2014) Molecular and cellular mechanisms responsible for cellular stress and low-grade inflammation induced by a super-low dose of endotoxin. J Biol Chem 289:16262–16269
Baker B et al (2015) Alteration of lysosome fusion and low-grade inflammation mediated by super-low-dose endotoxin. J Biol Chem 290:6670–6678
Ballesteros I et al (2020) Co-option of neutrophil fates by tissue environments. Cell 183:1282–1297.e1218
Bayne LJ et al (2012) Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 21:822–835
Bekkering S et al (2014) Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler Thromb Vasc Biol 34:1731–1738
Bhan C, Dipankar P, Chakraborty P, Sarangi PP (2016) Role of cellular events in the pathophysiology of sepsis. Inflamm Res 65:853–868
Biswas SK, Lopez-Collazo E (2009) Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol 30:475–487
Brady J, Horie S, Laffey JG (2020) Role of the adaptive immune response in sepsis. Intensive Care Med Exp 8:20
Buffen K et al (2014) Autophagy controls BCG-induced trained immunity and the response to intravesical BCG therapy for bladder cancer. PLoS Pathog 10:e1004485
Carnevale R et al (2018) Localization of lipopolysaccharide from Escherichia Coli into human atherosclerotic plaque. Sci Rep 8:3598
Casbon AJ et al (2015) Invasive breast cancer reprograms early myeloid differentiation in the bone marrow to generate immunosuppressive neutrophils. Proc Natl Acad Sci U S A 112:E566–E575
Chan C, Li L, McCall CE, Yoza BK (2005) Endotoxin tolerance disrupts chromatin remodeling and NF-kappaB transactivation at the IL-1beta promoter. J Immunol 175:461–468
Cheng Z, Taylor B, Ourthiague DR, Hoffmann A (2015) Distinct single-cell signaling characteristics are conferred by the MyD88 and TRIF pathways during TLR4 activation. Sci Signal 8:ra69
Choi YJ et al (2017) Impaired polyfunctionality of CD8(+) T cells in severe sepsis patients with human cytomegalovirus reactivation. Exp Mol Med 49:e382
Cortez-Retamozo V et al (2013) Angiotensin II drives the production of tumor-promoting macrophages. Immunity 38:296–308
De Santa F et al (2009) Jmjd3 contributes to the control of gene expression in LPS-activated macrophages. EMBO J 28:3341–3352
Deng H, Maitra U, Morris M, Li L (2013) Molecular mechanism responsible for the priming of macrophage activation. J Biol Chem 288:3897–3906
Efron PA et al (2018) Persistent inflammation, immunosuppression, and catabolism and the development of chronic critical illness after surgery. Surgery 164:178–184
Ene CI et al (2012) Histone demethylase Jumonji D3 (JMJD3) as a tumor suppressor by regulating p53 protein nuclear stabilization. PLoS One 7:e51407
Eruslanov EB et al (2014) Tumor-associated neutrophils stimulate T cell responses in early-stage human lung cancer. J Clin Invest 124:5466–5480
Foster SL, Hargreaves DC, Medzhitov R (2007) Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature 447:972–978
Frazier TH, DiBaise JK, McClain CJ (2011) Gut microbiota, intestinal permeability, obesity-induced inflammation, and liver injury. JPEN J Parenter Enteral Nutr 35:14S–20S
Fridlender ZG, Albelda SM (2012) Tumor-associated neutrophils: friend or foe? Carcinogenesis 33:949–955
Fu Y et al (2012) Network topologies and dynamics leading to endotoxin tolerance and priming in innate immune cells. PLoS Comput Biol 8:e1002526
Geng S et al (2016) The persistence of low-grade inflammatory monocytes contributes to aggravated atherosclerosis. Nat Commun 7:13436
Geng S, Zhang Y, Lee C, Li L (2019) Novel reprogramming of neutrophils modulates inflammation resolution during atherosclerosis. Sci Adv 5:eaav2309
Granot Z (2019) Neutrophils as a therapeutic target in cancer. Front Immunol 10:1710
Heidecke CD et al (1999) Selective defects of T lymphocyte function in patients with lethal intraabdominal infection. Am J Surg 178:288–292
Hirsh M, Mahamid E, Bashenko Y, Hirsh I, Krausz MM (2001) Overexpression of the high-affinity Fcgamma receptor (CD64) is associated with leukocyte dysfunction in sepsis. Shock 16:102–108
Hoechst B et al (2008) A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology 135:234–243
Holtzhausen A et al (2019) TAM family receptor kinase inhibition reverses MDSC-mediated suppression and augments anti-PD-1 therapy in melanoma. Cancer Immunol Res 7:1672–1686
Hong T, Xing J, Li L, Tyson J (2011) A mathematical model for the reciprocal differentiation of T helper 17 cells and induced regulatory T cells. PLoS Comput Biol 7:e1002122
Hong T, Xing J, Li L, Tyson JJ (2012) A simple theoretical framework for understanding heterogeneous differentiation of CD4+ T cells. BMC Syst Biol 6:66
Horiguchi H et al (2018) Innate immunity in the persistent inflammation, immunosuppression, and catabolism syndrome and its implications for therapy. Front Immunol 9:595
Hotchkiss RS et al (2016) Sepsis and septic shock. Nat Rev Dis Primers 2:16045
Jongstra-Bilen J et al (2006) Low-grade chronic inflammation in regions of the normal mouse arterial intima predisposed to atherosclerosis. J Exp Med 203:2073–2083
Kadelka S, Boribong BP, Li L, Ciupe SM (2019) Modeling the bistable dynamics of the innate immune system. Bull Math Biol 81:256–276
Kalafati L et al (2020) Innate immune training of granulopoiesis promotes anti-tumor activity. Cell 183:771–785.e712
Katsumata N et al (1996) Serum levels of cytokines in patients with untreated primary lung cancer. Clin Cancer Res 2:553–559
Kawai T, Akira S (2007) TLR signaling. Semin Immunol 19:24–32
Kishimoto T et al (2019) Serum levels of the chemokine CCL2 are elevated in malignant pleural mesothelioma patients. BMC Cancer 19:1204
Kiss M, Caro AA, Raes G, Laoui D (2020) Systemic reprogramming of monocytes in cancer. Front Oncol 10:1399
Kobayashi M et al (2019) Blocking monocytic myeloid-derived suppressor cell function via anti-DC-HIL/GPNMB antibody restores the in vitro integrity of T cells from cancer patients. Clin Cancer Res 25:828–838
Kovach MA, Standiford TJ (2012) The function of neutrophils in sepsis. Curr Opin Infect Dis 25:321–327
Kumar V (2020) Understanding the complexities of SARS-CoV2 infection and its immunology: a road to immune-based therapeutics. Int Immunopharmacol 88:106980
Kumar V (2021a) Emerging human coronavirus infections (SARS, MERS, and COVID-19): where they are leading us. Int Rev Immunol 40:5–53
Kumar V (2021b) How could we forget immunometabolism in SARS-CoV2 infection or COVID-19? Int Rev Immunol 40:72–107
Laird MH et al (2009) TLR4/MyD88/PI3K interactions regulate TLR4 signaling. J Leukoc Biol 85:966–977
Lassenius MI et al (2011) Bacterial endotoxin activity in human serum is associated with dyslipidemia, insulin resistance, obesity, and chronic inflammation. Diabetes Care 34:1809–1815
Lerias JR et al (2019) Trained immunity for personalized cancer immunotherapy: current knowledge and future opportunities. Front Microbiol 10:2924
Li J, Lin S, Vanhoutte PM, Woo CW, Xu A (2016) Akkermansia muciniphila protects against atherosclerosis by preventing metabolic endotoxemia-induced inflammation in Apoe−/− mice. Circulation 133:2434–2446
Li L, McCall C, Hu X (2020) Editorial: innate immunity programming and memory in resolving and non-resolving inflammation. Front Immunol 11:177
Libby P, Hansson GK (2015) Inflammation and immunity in diseases of the arterial tree: players and layers. Circ Res 116:307–311
Libby P et al (2019) Atherosclerosis. Nat Rev Dis Primers 5:56
Lin R, Zhang Y, Pradhan K, Li L (2020) TICAM2-related pathway mediates neutrophil exhaustion. Sci Rep 10:14397
Lissauer ME et al (2009) Differential expression of toll-like receptor genes: sepsis compared with sterile inflammation 1 day before sepsis diagnosis. Shock 31:238–244
Liu Q, Li CS (2017) Programmed cell death-1/programmed death-ligand 1 pathway: a new target for sepsis. Chin Med J (Engl) 130:986–992
Lopez-Collazo E, Avendano-Ortiz J, Martin-Quiros A, Aguirre LA (2020) Immune response and COVID-19: a mirror image of sepsis. Int J Biol Sci 16:2479–2489
Lu G et al (2015) Myeloid cell-derived inducible nitric oxide synthase suppresses M1 macrophage polarization. Nat Commun 6:6676
Luczynski W, Stasiak-Barmuta A, Krawczuk-Rybak M (2004) Lower percentages of monocytes with CD80, CD86 and HLA-DR molecule expression in pediatric cancer. Cancer Immunol Immunother 53:1049–1050
Lundberg AM et al (2013) Toll-like receptor 3 and 4 signalling through the TRIF and TRAM adaptors in haematopoietic cells promotes atherosclerosis. Cardiovasc Res 99:364–373
Maitra U et al (2012) Molecular mechanisms responsible for the selective and low-grade induction of proinflammatory mediators in murine macrophages by lipopolysaccharide. J Immunol 189:1014–1023
Manz MG, Boettcher S (2014) Emergency granulopoiesis. Nat Rev Immunol 14:302–314
Morris MC, Gilliam EA, Li L (2014) Innate immune programing by endotoxin and its pathological consequences. Front Immunol 5:680
Nathan C, Ding A (2010) Nonresolving inflammation. Cell 140:871–882
Netea MG et al (2016) Trained immunity: a program of innate immune memory in health and disease. Science 352:aaf1098
Netea MG, Joosten LAB, van der Meer JWM (2017) Hypothesis: stimulation of trained immunity as adjunctive immunotherapy in cancer. J Leukoc Biol 102:1323–1332
Niu B et al (2019) Different expression characteristics of LAG3 and PD-1 in Sepsis and their synergistic effect on T cell exhaustion: a new strategy for immune checkpoint blockade. Front Immunol 10:1888
Nywening TM et al (2016) Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol 17:651–662
O’Sullivan ST et al (1995) Major injury leads to predominance of the T helper-2 lymphocyte phenotype and diminished interleukin-12 production associated with decreased resistance to infection. Ann Surg 222:482–490.; discussion 490–482
Olingy CE, Dinh HQ, Hedrick CC (2019) Monocyte heterogeneity and functions in cancer. J Leukoc Biol 106:309–322
Opal SM et al (2013) Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA 309:1154–1162
Otto GP et al (2011) The late phase of sepsis is characterized by an increased microbiological burden and death rate. Crit Care 15:R183
Palsson-McDermott EM, O'Neill LA (2004) Signal transduction by the lipopolysaccharide receptor, toll-like receptor-4. Immunology 113:153–162
Patil NK, Guo Y, Luan L, Sherwood ER (2017) Targeting immune cell checkpoints during sepsis. Int J Mol Sci 18
Paula-Neto HA et al (2011) Inhibition of guanylyl cyclase restores neutrophil migration and maintains bactericidal activity increasing survival in sepsis. Shock 35:17–27
Perner A et al (2016) Sepsis: frontiers in diagnosis, resuscitation and antibiotic therapy. Intensive Care Med 42:1958–1969
Piao W et al (2009) Endotoxin tolerance dysregulates MyD88- and toll/IL-1R domain-containing adapter inducing IFN-beta-dependent pathways and increases expression of negative regulators of TLR signaling. J Leukoc Biol 86:863–875
Poschke I, Mougiakakos D, Hansson J, Masucci GV, Kiessling R (2010) Immature immunosuppressive CD14+HLA-DR-/low cells in melanoma patients are Stat3hi and overexpress CD80, CD83, and DC-sign. Cancer Res 70:4335–4345
Powell DR, Huttenlocher A (2016) Neutrophils in the tumor microenvironment. Trends Immunol 37:41–52
Rahtes A, Li L (2020) Polarization of low-grade inflammatory monocytes through TRAM-mediated up-regulation of Keap1 by super-low dose endotoxin. Front Immunol 11:1478
Rhee C et al (2019) Prevalence, underlying causes, and preventability of sepsis-associated mortality in US acute care hospitals. JAMA Netw Open 2:e187571
Ribechini E et al (2017) Novel GM-CSF signals via IFN-gammaR/IRF-1 and AKT/mTOR license monocytes for suppressor function. Blood Adv 1:947–960
Rosenbloom AJ et al (1999) Suppression of cytokine-mediated beta2-integrin activation on circulating neutrophils in critically ill patients. J Leukoc Biol 66:83–89
Rudd KE et al (2020) Global, regional, and national sepsis incidence and mortality, 1990-2017: analysis for the global burden of disease study. Lancet 395:200–211
Ryan T, Coakley JD, Martin-Loeches I (2017) Defects in innate and adaptive immunity in patients with sepsis and health care associated infection. Ann Transl Med 5:447
Scholl SM et al (1996) Circulating levels of the macrophage colony stimulating factor CSF-1 in primary and metastatic breast cancer patients. A pilot study. Breast Cancer Res Treat 39:275–283
Schulte-Schrepping J et al (2020) Severe COVID-19 is marked by a dysregulated myeloid cell compartment. Cell 182:1419–1440.e1423
Serhan CN, Savill J (2005) Resolution of inflammation: the beginning programs the end. Nat Immunol 6:1191–1197
Shao R et al (2016) Monocyte programmed death ligand-1 expression after 3-4 days of sepsis is associated with risk stratification and mortality in septic patients: a prospective cohort study. Crit Care 20:124
Shen XF, Cao K, Jiang JP, Guan WX, Du JF (2017) Neutrophil dysregulation during sepsis: an overview and update. J Cell Mol Med 21:1687–1697
Singhal S et al (2016) Origin and role of a subset of tumor-associated neutrophils with antigen-presenting cell features in early-stage human lung cancer. Cancer Cell 30:120–135
Sinistro A et al (2008) Downregulation of CD40 ligand response in monocytes from sepsis patients. Clin Vaccine Immunol 15:1851–1858
Steggerda SM et al (2017) Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J Immunother Cancer 5:101
Stoll LL, Denning GM, Weintraub NL (2004) Potential role of endotoxin as a proinflammatory mediator of atherosclerosis. Arterioscler Thromb Vasc Biol 24:2227–2236
Stoppacciaro A et al (1993) Regression of an established tumor genetically modified to release granulocyte colony-stimulating factor requires granulocyte-T cell cooperation and T cell-produced interferon gamma. J Exp Med 178:151–161
Strauss L et al (2020) Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci Immunol 5
Sugimoto K et al (2003) Monocyte CD40 expression in severe sepsis. Shock 19:24–27
Trovato R et al (2019) Immunosuppression by monocytic myeloid-derived suppressor cells in patients with pancreatic ductal carcinoma is orchestrated by STAT3. J Immunother Cancer 7:255
Ugurel S et al (2004) Down-regulation of HLA class II and costimulatory CD86/B7-2 on circulating monocytes from melanoma patients. Cancer Immunol Immunother 53:551–559
Wang JF et al (2015) Up-regulation of programmed cell death 1 ligand 1 on neutrophils may be involved in sepsis-induced immunosuppression: an animal study and a prospective case-control study. Anesthesiology 122:852–863
Weber J et al (2016) Phase I/II study of metastatic melanoma patients treated with nivolumab who had progressed after Ipilimumab. Cancer Immunol Res 4:345–353
Weide B et al (2014) Myeloid-derived suppressor cells predict survival of patients with advanced melanoma: comparison with regulatory T cells and NY-ESO-1- or melan-A-specific T cells. Clin Cancer Res 20:1601–1609
Wherry EJ (2011) T cell exhaustion. Nat Immunol 12:492–499
Wick M, Kollig E, Muhr G, Koller M (2000) The potential pattern of circulating lymphocytes TH1/TH2 is not altered after multiple injuries. Arch Surg 135:1309–1314
Wiersinga WJ et al (2009) Immunosuppression associated with interleukin-1R-associated-kinase-M upregulation predicts mortality in gram-negative sepsis (melioidosis). Crit Care Med 37:569–576
Wiesner P et al (2010) Low doses of lipopolysaccharide and minimally oxidized low-density lipoprotein cooperatively activate macrophages via nuclear factor kappa B and activator protein-1: possible mechanism for acceleration of atherosclerosis by subclinical endotoxemia. Circ Res 107:56–65
Wu WC et al (2014) Circulating hematopoietic stem and progenitor cells are myeloid-biased in cancer patients. Proc Natl Acad Sci U S A 111:4221–4226
Xiong Y, Medvedev AE (2011) Induction of endotoxin tolerance in vivo inhibits activation of IRAK4 and increases negative regulators IRAK-M, SHIP-1, and A20. J Leukoc Biol 90:1141–1148
Yang L et al (2019) Histone demethylase KDM6B has an anti-tumorigenic function in neuroblastoma by promoting differentiation. Oncogenesis 8:3
Yuan R et al (2016a) Low-grade inflammatory polarization of monocytes impairs wound healing. J Pathol 238:571–583
Yuan R, Geng S, Li L (2016b) Molecular mechanisms that underlie the dynamic adaptation of innate monocyte memory to varying stimulant strength of TLR ligands. Front Immunol 7:497
Zhang Y, Lee C, Geng S, Li L (2019) Enhanced tumor immune surveillance through neutrophil reprogramming due to Tollip deficiency. JCI Insight 4:e122939
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Geng, S., Pradhan, K., Li, L. (2021). Signal-Strength and History-Dependent Innate Immune Memory Dynamics in Health and Disease. In: Kumar, V. (eds) Toll-like Receptors in Health and Disease. Handbook of Experimental Pharmacology, vol 276. Springer, Cham. https://doi.org/10.1007/164_2021_485
Download citation
DOI: https://doi.org/10.1007/164_2021_485
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-06511-8
Online ISBN: 978-3-031-06512-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)