
Abstract
Farnesoid X receptor controls bile acid metabolism, both in the liver and intestine. This potent nuclear receptor not only maintains homeostasis of its own ligands, i.e., bile acids, but also regulates glucose and lipid metabolism as well as the immune system. These findings have led to substantial interest for FXR as a therapeutic target and to the recent approval of an FXR agonist for treating primary biliary cholangitis as well as ongoing clinical trials for other liver diseases. Given that FXR biology is complex, including moderate expression in tissues outside of the enterohepatic circulation, temporal expression of isoforms, posttranscriptional modifications, and the existence of several other bile acid-responsive receptors such as TGR5, clinical application of FXR modulators warrants thorough understanding of its actions. Recent findings have demonstrated remarkable physiological effects of targeting FXR specifically in the intestine (iFXR), thereby avoiding systemic release of modulators. These include local effects such as improvement of intestinal barrier function and intestinal cholesterol turnover, as well as systemic effects such as improvements in glucose homeostasis, insulin sensitivity, and nonalcoholic fatty liver disease (NAFLD). Intriguingly, metabolic improvements have been observed with both an iFXR agonist that leads to production of enteric Fgf15 and increased energy expenditure in adipose tissues and antagonists by reducing systemic ceramide levels and hepatic glucose production. Here we review the recent findings on the role of intestinal FXR and its targeting in metabolic disease.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
The bile acid-activated farnesoid X receptor (FXR, NR1H4) is a major regulator of bile acid homeostasis and is, accordingly, most highly expressed in liver and intestine, i.e., the organs that physically constitute the enterohepatic circulation of bile acids. FXR belongs to the nuclear receptor family of ligand-activated transcription factors that are activated by hydrophobic molecules such as steroids, hormones, and fatty acids. Upon their activation these receptors control a wide array of processes in health and disease and, importantly, can be specifically targeted by pharmacological means. In fact, approximately 13% of all FDA-approved drugs target members of the nuclear receptor family (Overington et al. 2006). The identification of FXR as a bile acid-activated nuclear receptor in 1999 (Makishima et al. 1999; Parks et al. 1999; Wang et al. 1999), and the subsequent discovery of TGR5 (GPBAR1) as a bile acid-activated membrane-bound G protein-coupled receptor (Maruyama et al. 2002), has led to the identification of a series of novel physiological functions for bile acids that go far beyond their “classic” roles in the generation of bile flow and facilitating the uptake of dietary fats and fat-soluble vitamins (Hofmann and Hagey 2014; Kuipers et al. 2014). These novel functions in the regulation of glucose and lipid metabolism and in modulation of inflammation have raised substantial interest in designing therapeutic approaches that are based on interference with the bile acid-sensing machinery. Already in 2016, the FXR agonist obeticholic acid (OCA, Ocaliva also known as INT-747), an analogue of the natural FXR ligand chenodeoxycholic acid (CDCA), developed by Intercept Pharmaceuticals was the first treatment approved for primary biliary cholangitis (PBC) in 20 years and is currently under investigation in clinical trials for other liver diseases (Nevens et al. 2016). Fundamental studies on FXR have not only delineated a wide array of physiological functions for this nuclear receptor, but also pinpointed tissue-specific actions at its major sites of expression, the liver and intestine, as well as in other organs where expression is moderate yet with evident biological function. For instance, FXR activation in the kidney has nephroprotective effects (Herman-Edelstein et al. 2018), FXR modulates adipocyte differentiation in white adipose tissue (Abdelkarim et al. 2010), and it may even be involved in the etiology of depression through actions in the brain (Chen et al. 2018). In line with a “hormone-like function,” bile acid pool size and composition, the rate of intestinal absorption of the individual bile acid species, and the efficiency of hepatic uptake of these intestine-derived bile acids during their enterohepatic cycling are increasingly recognized as important metabolic cues. For instance, some of the (immediate) beneficial metabolic effects of bariatric surgery have been attributed to altered bile acid metabolism (Spinelli et al. 2016; Albaugh et al. 2018), in particular via FXR-dependent changes in microbiome composition (Ryan et al. 2014). Yet, the exact contribution hereof and their underlying mechanisms, including a potential specific role of iFXR, remain to be elucidated.
FXR biology appears to be complex. Apart from its widespread organ and tissue expression, there are multiple FXR isoforms, posttranslational modifications, and regulatory cofactors that eventually all contribute to FXR activity. Therefore, meaningful pharmacological manipulation of FXR activity for therapeutic purposes is also complex but, at the same time, offers great opportunities for pursuit of novel strategies to develop selective modulators in a tissue- or function-specific manner. This has collectively been designated as the search for selective bile acid receptor modulators (SBARMs, as discussed by Massafra et al. 2018). This is not an easy task as, for instance, mice treated with the FXR ligand GW4064 showed binding of activated FXR to 6,345 binding sites on the genome in the liver upon ChipSeq analysis yet to 3,872 other sites in the intestine and to 1,449 joint sites in both organs. These sites were reported to change dramatically in obesity, illustrating both the complexity and the great potential of FXR modulation (Thomas et al. 2009 ; Lee et al. 2012).
The intestine is an attractive candidate for tissue-specific FXR modulation, as therapeutics can be administered orally, and, if bioavailability can be limited to the gut, systemic release is avoided and side effects potentially prevented. At the same time, intestine-derived factors released upon treatment can exert systemic beneficial effects. Tissue-specific intestinal FXR (iFXR) modulators indeed appear to induce systemic metabolic improvements in obese mice, through actions in adipose tissue and liver (Fang et al. 2015; Pathak et al. 2018). However, these effects were paradoxically shown to occur with both activation and inhibition of iFXR, indicating that modulating the interplay of iFXR with its surroundings is not as straightforward as theoretically contemplated (Jiang et al. 2015a; Fang et al. 2015).
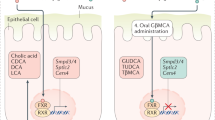
This review will focus on the physiological consequences of pharmacological modulation of iFXR. First, the characteristics and physiological function of endogenous iFXR will be outlined, starting with its role in modulation of bile acid homeostasis, since this aspect has to be taken into consideration when one plans to interfere with iFXR activity (Fig. 1). Recent findings on the physiological effects of selectively manipulating iFXR in pathophysiological settings will be discussed in the context of prospective applications of this promising therapeutic target.

Overview of intestinal FXR signaling. Following ingestion of a meal, conjugated bile acids are released from the gallbladder into the duodenum where they form mixed micelles that aid in the digestion of fats and fat-soluble vitamins. More distally, BAs can be deconjugated and converted into secondary bile acids by intestinal microbiota or/and transported into enterocytes via ASBT in the ileum. Upon bile acid uptake, iFXR can be activated, leading to transcription of SHP, FGF15/19, and OSTA/B, among others. Conjugated muricholic acids antagonize FXR activation in mice, and glycoursodeoxycholic acid (GUDCA) may do so in humans. At the basolateral side of the enterocytes, bile acids are transported by OSTα/β into portal blood. FGF15/19 is also secreted basolaterally, and both classes of compounds are transported to the liver. Upon reaching the liver, FGF15/19 inhibits hepatic bile acid synthesis. Activation of iFXR and FGF15/19 decreases hydrophobicity of the bile acid pool that stimulates transintestinal cholesterol excretion, predominantly upstream of the ileum. Bile acids stimulate TGR5 in the apical membrane of intestinal L cells, leading to release of glucagon-like peptide (GLP1), a process that appears to be modulated by FXR. Importantly, GCG expression, encoding preproglucagon and ultimately GLP1, is also suppressed by FXR. FXR activation furthermore maintains intestinal barrier function by stimulation of tight junction formation and suppressing inflammation in immune cells. Both GLP-1 and FGF15/19 have distinct systemic effects by activating their respective receptors in various organs
1.1 Role of iFXR in Control of Bile Acid Metabolism
Because bile acids are “natural detergents”, their hepatic synthesis rate, transport across cell membranes, and circulating pool size need to be tightly regulated, to ensure optimal concentrations in the intestinal lumen to facilitate nutrient absorption and, at the same time, prevent cytotoxicity at the sites where bile acids accumulate. Besides in hepatocytes (Goodwin et al. 2000), FXR in the distal ileum also contributes to the regulation of hepatic bile acid synthesis upon activation by circulating bile acids, thereby completing an effective negative feedback loop. In fact, under physiological conditions, signaling through iFXR appears to dominate de novo bile acid synthesis in rodents (Inagaki et al. 2005; Kim et al. 2007), and this regulation appears to be conserved in humans (Sjöberg et al. 2017). In short, the primary bile acids cholic acid (CA, 3α,7α,12α-trihydroxy-5β-cholan-24-oic acid) and CDCA (3α,7α-dihydroxy-5β-cholan-24-oic acid) are synthesized from cholesterol and conjugated to either taurine or glycine before their secretion into bile via the bile salt export pump (BSEP, ABCB11). In rodents the primary bile acids α- and β-muricholic acid (3α,6β,7α-trihydroxy-5β-cholan-24-oic acid and 3α,6β,7β-trihydroxy-5β-cholan-24-oic acid, respectively) are also synthesized. Upon secretion into the intestine, primary bile acids can be deconjugated by gut microbial bile salt hydrolases (BSH) and subsequently converted into secondary bile acids by bacterial 7α-dehydroxylases, leading to formation of deoxycholic acid (DCA) from CA and lithocholic acid (LCA) from CDCA. Likely due to the high prevailing bile acid concentrations, the microbial content is the lowest in the proximal part of the small intestine (103/g in duodenum) and gradually increases (107/g in ileum) toward the large intestine, where microbial counts are the highest (1012/g in colon) (Mowat and Agace 2014). The capacity to deconjugate and dehydroxylate bile acids, consequently, increases along the intestinal tract. Bile acid deconjugation, which occurs largely in the distal part of the small intestine, is required for passive diffusion across the intestinal membrane. Active transport by the apical sodium-bile acid transporter (ASBT) is also confined to the ileum. This ecosystem ensures a sufficiently high bile acid concentration in the upper intestine to enable fat absorption. CDCA can be epimerized at the C7 position by some bacterial species to yield ursodeoxycholic acid (UDCA), a relatively hydrophilic bile acid (Fedorowski et al. 1979). Thus, there is a mutual interaction between the microbiome and bile acids, which might benefit bile acid-metabolizing strains (Friedman et al. 2018). Each bile acid species with its specific number and orientation of hydroxyl groups and, hence, hydrophobicity (Heuman 1989) differs in its ability to activate FXR (and TGR5). Therefore, local bile acid concentrations as well as composition will determine ensuing physiological responses. CDCA has been recognized as the most potent endogenous ligand of FXR, followed by DCA, LCA, and CA, whereas TGR5 is most potently activated by LCA, followed by DCA (Pathak et al. 2017). In contrast, TβMCA is a natural FXR antagonist in mice (Mueller et al. 2015; Sayin et al. 2013), while glycoursodeoxycholic acid (GUDCA) was recently reported to act as an FXR antagonist in humans (Sun et al. 2018a). LCA is the most hydrophobic bile acid species present in the adult human bile acid pool and is considered the most toxic (i.e., cholestatic), whereas the hydrophilic UDCA strongly stimulates bile flow (i.e., choleretic) and is being used in the treatment of cholestatic conditions such as PBC and intrahepatic cholestasis of pregnancy (Lefebvre et al. 2009; Cariello et al. 2018).
Bile acids are excreted from ileocytes (i.e., ileal enterocytes), into the blood by the basolaterally localized transporters OSTα/β and subsequently travel to the liver. After hepatic uptake by the basolateral transporter NTCP (SLC10A1), intracellular bile acids can activate hepatic FXR to regulate bile acid synthesis by inducing small heterodimer partner (SHP, NR0B2), which suppresses the expression of CYP7A1 and CYP8B1 (cholesterol 7 alpha-hydroxylase and sterol 12-alpha-hydroxylase respectively), encoding key enzymes in the conversion of cholesterol to bile acids (Lu et al. 2000; Goodwin et al. 2000). Importantly, iFXR induces production of fibroblast growth factor 19 (FGF19) in humans and Fgf15 in mice that also contributes to the regulation of hepatic bile acid synthesis (Kim et al. 2007). Composition, and hence hydrophobicity, of the bile acid pool is an important determinant of several physiological functions of circulating bile acids. This hydrophobicity is determined by the ratio in which the distinct primary bile acids are being formed as well as by their conversion into secondary species in the gut. For example, stimulation of iFXR in mice with the selective FXR agonist PX20606 leads to a very hydrophilic bile acid pool in mice, consisting almost exclusively of muricholic acids, that drives increased excretion of cholesterol from enterocytes into the intestinal lumen, mostly in the duodenum (described below, de Boer et al. 2017). Importantly, iFXR also contributes to control of (postprandial) gallbladder filling, through FGF15/19-induced relaxation of the gallbladder smooth muscle, hence modulating the dynamics of the enterohepatic circulation of bile acids (Choi et al. 2006).
Of note, the nuclear receptors pregnane X receptor (PXR; NR1I2), vitamin D receptor (VDR; Nr1I1), constitutive androstane receptor (CAR; NR1I3), as well as the sphingosine 1-phosphate receptor 2 (S1PR2) are all also activated by bile acids (recently reviewed by Shapiro et al. 2018; Massafra et al. 2018). As activation of FXR modulates bile acid composition as well as bile acid concentrations in the various body compartments, manipulating FXR activity inevitably also affects the activities of the other bile acid-responsive receptors.
A large variation in bile acid composition is observed in the general human population (e.g., Luo et al. 2018), reflecting the interplay between bile acid sensors and factors such as lifestyle, diet, and the microbiome in the maintenance of bile acid homeostasis. Furthermore, various disease states are characterized by altered bile acid composition: this does not only concern liver diseases (Armstrong and Guo 2017; Trauner et al. 2017) but also cystic fibrosis (Bertolini et al. 2019) and metabolic diseases such as type 2 diabetes (e.g., Brufau et al. 2010). This widely varying “endogenous FXR activating capacity” has to be taken into account in the development of new therapeutic strategies based on interference with FXR activity rather than assuming that a one-size-fits-all procedure will be successful. Along the same line, it is evident that preclinical studies in murine models are of limited translational value because of the presence of large amounts of hydrophilic muricholates with antagonistic actions. Therefore mouse models with a humanized bile acid pool are urgently needed.
2 Intestinal FXR Isoforms and Posttranscriptional Regulation
The majority of intestinal bile acids is taken up via ASBT in the terminal ileum, leading to activation of FXR in ileocytes. However, FXR is expressed along the entire length of the small intestine as well as the colon (as deduced from human and mouse literature using Genevestigator analysis, data not shown, Zimmermann et al. 2004). Yet, the physiological role of FXR in the proximal part of the gastrointestinal tract, and its potential ligands, is still ill-defined. Next to these local distribution patterns, there are additional levels of regulation that may impact on physiological outcome of FXR activation in the various parts of the intestine. First, it is well known that multiple isoforms of FXR exist in humans and in rodents, with differences in spatiotemporal expression patterns, and that FXR is subject to posttranslational modification. These aspects may all contribute to fine-tuning of iFXR activity and will be further discussed in the next paragraphs.
2.1 Farnesoid X Receptor Isoforms
Human FXRα (or NR1H4) is expressed from a single gene locus located on chromosome 12 (12q23.1) and murine FXR on chromosome 10 (10:89454234–89533585). Differential promoter regulation and alternative splicing result in four different isoforms (FXRα1, FXRα2, FXRα3, and FXRα4) both in humans and in mice (Zhang et al. 2003; Huber et al. 2002). Two promoters, present in front of exon 1 and 3, induce either expression of FXRα1,2 or α3,4 (Fig. 2a). A point of caution, generally overlooked, is that public databases (NCBI, ENSEMBL) only contain three RefSeq transcripts of mouse FXR, missing the α2 isoform. Moreover two additional splice variants of the human α1 isoform are reported that encode the same protein. Besides FXRα, mice have a functional pseudogene FXRβ, which appears nonfunctional in primates, and that is activated by the cholesterol synthesis intermediate lanosterol (Otte et al. 2003). FXRβ will not be considered in this chapter.

(a) The farnesoid X receptor gene structure and predicted isoforms. (b) FXR protein domains and reported sites for modifications by acetylation, phosphorylation, SUMOylation, and O-GlcNAcylation as detailed in the text
Expression of human FXR is highest in ileum, followed by liver, duodenum, kidney, and colon (Vaquero et al. 2013). Genevestigator database analysis showed a similar order in mice, with the exception of a relatively high Fxr expression in the kidney, comparable to that in the liver (data not shown, Zimmermann et al. 2004; Boesjes et al. 2014). FXRα1–4 isoforms were reported to be equally expressed in the liver, while FXRα3 and FXRα4 predominate in duodenum, jejunum, and ileum (Zhang et al. 2003; Boesjes et al. 2014). Regulation of FXR target genes is reported mostly in an isoform-independent manner; however, some genes (e.g., IBABP) appear to be more responsive to FXRα2/FXRα4 than to FXRα1/FXRα3, due to the lack of the additional amino acids next to the DNA-binding domain (MYTG motif) (Zhang et al. 2003). The lack of this MYTG motif in FXRα2 and FXRα4 in general results in higher transcriptional activity (Gray and Squires 2015). The presence of the tyrosine phosphorylation site in the MYTG motif was suggested to be responsible for differences in variant activity (Gray and Squires 2015). Whether MYTG is increasing or decreasing the activity of FXR seems to be species-dependent. In mice, hepatic FXRα2 was proven to be more effective than FXRα4 in reducing HDL- and VLDL-cholesterol levels and in switching hydrophobicity of the bile acid pool due to differential regulation of Cyp8b1 expression. In addition, hepatic FXRα2 caused an increased fecal neutral sterol excretion without affecting intestinal cholesterol absorption when compared to hepatic FXRα4 (Boesjes et al. 2014). Furthermore, a cell-specific pattern of FXR isoforms seems to determine the tissue sensitivity to FXR agonists, which may also be specific for different target genes (Zhang et al. 2003; Vaquero et al. 2013). Selective effects of the different isoforms specifically on iFXR targets have to the best of our knowledge not been reported.
The transcription factor GATA binding protein 4 (GATA4) suppresses bile acid metabolism-related genes in the jejunum: this regulator of intestinal development defines jejunal versus ileal identity (Walker et al. 2014; Thompson et al. 2017). Disruption of the microbiota with antibiotics resulted in jejunal expression of Asbt in a Gata4-dependent manner that confines Asbt expression to the ileal part of the intestine (Out et al. 2015). Bile acid-mediated activation of FXR in the small intestine therefore appears to be mainly restricted to terminal ileum by an interaction between GATA4 and the microbiota (Out et al. 2015).
2.2 Posttranscriptional Regulation of FXR by Phosphorylation, Acetylation, O-GlcNAcylation, or SUMOylation
Posttranslational regulation of FXR has been reported and has been related to various metabolic changes. These modulations can have both repressing and activating effects on FXR function (summarized in Fig. 2b). While most of these interactions with protein-modifying enzymes have been described for hepatic FXR, these enzymes are also present in the intestine, and they are therefore likely to act accordingly in enterocytes. In the next sections, we will discuss the different posttranslational modifications of FXR that need to be taken into account when studying the impact of pharmacological iFXR modulation, particularly in metabolic disease states.
Protein kinase C (PKC) has been shown to phosphorylate FXR at serine (S)135 and S154 in the DNA-binding domain, leading to recruitment of coactivator PGC1α and enhanced FXR transcriptional activity (Zhang et al. 2004; Gineste et al. 2008). In contrast, phosphorylation of S250 by AMPK, one of the main sensors of cellular energy levels, leads to repression of FXR, which, when left uncontrolled, leads to bile acid accumulation and hepatic injury (Lien et al. 2014; Becares et al. 2017). Phosphorylation of FXR by PKC ζ that is stimulated by ATP8B1 was shown to be one of the determinants of translocation to the nucleus (Frankenberg et al. 2008). Nuclear translocation was later shown to apply to all isoforms of FXR in a tissue- and species-independent manner (Vaquero et al. 2013). As a result, probably due to insufficient capacity of BSEP, accumulation of hepatic bile acids occurs when the phospholipid-transporting ATPase ATP8B1 does not function properly. Mutations in ATP8B1 cause progressive familial intrahepatic cholestasis type 1 (PFIC1, Bull et al. 1998). As extrahepatic manifestations of mutations in ATP8B1 occur as well, including diarrhea that often does not resolve after liver transplantation, a similar regulation of FXR activity by ATP8B1/PKC-induced phosphorylation may occur in enterocytes. However, ATP8B1 encodes for a phospholipid flippase, which may primarily affect the endothelial membrane and perhaps have secondary effects on FXR. Yet, regulation of apical localization of proteins in endothelial cells by ATP8B1 was shown to be independent from its flippase activity (Verhulst et al. 2010). Direct evidence of FXR phosphorylation in the intestine has not been reported (Frankenberg et al. 2008).
FXR was shown to control expression of miR-34a and its target (among others) sirtuin 1 (SIRT1), a mediator of the beneficial effects of caloric restriction in rodents (Lee and Kemper 2010). The activity of several transcription factors involved in regulation of metabolic genes, including FXR, can in turn be altered by SIRT1-mediated deacetylation. Interestingly acetylated FXR levels are remarkably increased in fatty livers of obese mice, suggesting a decreased activity of SIRT1. Indeed, administration of SIRT1 agonists to these obese mice decreased FXR acetylation and had beneficial metabolic effects (Kemper et al. 2009). In addition, both acetylation of K157 and K217 were shown to stabilize FXR, coinciding with a decreased capability to heterodimerize with retinoid X receptor (RXR) (Kemper et al. 2009).
Increased acetylation of FXR at K217 in obese mice was shown to be associated with inhibition of SUMO2 modification at the K277 position, resulting in increased hepatic inflammation and metabolic dysfunction (Kim et al. 2014). In addition, SUMOylation of K122 and K275 leads to decreased recruitment of FXR to target gene promotors and increased interaction with nuclear factor-kappa beta (NF-kB) (Balasubramaniyan et al. 2013). Recently, another human SUMOylation site was identified (i.e., L325) in an FXR domain that is responsible for transcriptional coactivation. Posttranslational modification of this site was required to achieve efficient ligand activation (Bilodeau et al. 2017). Whereas posttranslational modifications have been primarily reported for hepatic FXR, remarkably little is known about acetylation and SUMOylation of iFXR in the intestine, i.e., in an organ with high metabolic rate.
In liver, O-GlcNAcylation of FXR (S62 in the AF-1 domain) occurs in response to glucose and was shown to increase FXR protein levels and transcriptional activity (Berrabah et al. 2014). Pathophysiology in relation to FXR O-GlcNAcylation has not yet been reported, but it has been suggested that this modification might affect FXR-mediated control on bile acid production (Benhamed et al. 2015). Additionally, in vitro methylation of FXR was reported in hepatocytes on K206 (Balasubramaniyan et al. 2012), and in a large proteomic analysis, ubiquitylation of hepatic FXR was identified as well (Wagner et al. 2012). As conjugated and unconjugated BAs seem to differentially activate FXR in different cell lines, an as yet undefined posttranscriptional modification of FXR or the recruitment of different coactivators to the transcription complex was proposed (Vaquero et al. 2013). Thus, it seems that conjugated BAs require an additional cellular mechanism to activate FXR, which, although the exact mechanism remains ill-defined, might have physiological implication along the length of the small intestine where unconjugated bile acids can be taken up proximally by passive diffusion and conjugated ones only in the ileum by active ASBT-mediated transport.
2.3 FXR in Development and Aging
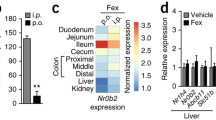
An intriguing yet poorly explored aspect of FXR biology concerns its potential role(s) during the different phases of life. It is well established that bile acid metabolism in the fetus and newborn shows specific features, in part due to the development of the newborn’s microbiome and in part dependent on the developmental pattern in the bile acid synthesis cascade, i.e., during the late fetal and early postnatal phase, a phase of physiologic cholestasis may occur that gives rise to increased ligand availability (Suchy et al. 1981 ; Stahl et al. 1993; Hill et al. 2017). Yet, surprisingly little is known about expression and activity of FXR during this period of continuous adaptive change. As a first step to gain a basic understanding of this period that is crucial for metabolic homeostasis later in life, we have performed a database search of iFxr expression in the intestine during the mouse life cycle (Genevestigator, Zimmermann et al. 2004), which revealed very high expression levels in utero that decrease somewhat after birth and remain relatively constant throughout life (Fig. 3a). To further specify iFxr expression in early life that was only represented by a single time point in the database analysis, we determined mRNA levels of FXR isoforms α1/2 and α3/4 in intestine and livers of developing mouse embryos and compared these to adult levels. Figure 3b shows that in intestine isoforms α3/4 dramatically increase from embryonic day 15 after pregnancy to day 19, while expression of α1/2 lags behind. Fetal expression in the intestine appeared to be higher than in adults. In contrast, in the fetal liver, isoforms α1/2 dominate over α3/4 and increase from day 15 until birth, but hepatic expression appears to be lower in the fetus than in the adult. It should be emphasized that the physiological relevance hereof awaits further study.

(a) Genevestigator analysis of intestinal Fxr (Nr1h4) expression during the murine life course, showing highest expression prior to birth. (b) Intestinal and hepatic Fxr isoform expression in utero, showing increasing expression from embryonic day 15 until birth with highest expression of Fxr α3/α4 in the intestine and predominating expression of the α1/α2 isoforms in the liver that is highest in adult mice
Postnatally, various metabolic changes occur that have been associated with age-related development of metabolic diseases. For example, decreased conversion of cholesterol into bile acids potentially contributes to risk of cardiovascular disease which is decreased in aging (Charach et al. 2017), implying changes in bile acid receptor activity (Uranga and Keller 2010; Joyce and Gahan 2016). Indeed, it has been shown that FXR expression in kidney and liver is decreased in aged mice (Xiong et al. 2014; Wang et al. 2017), whereas our analysis did not show this for iFxr (Fig. 3b). In contrast, aging of both whole-body and liver-specific Fxr/Shp double knockout mice showed reversal of body weight gain, adiposity, and glucose/insulin tolerance associated with aging (Kim et al. 2017). Intestine-specific FXR reactivation restores bile acid homeostasis in young and aged mice that lack FXR, which protects them from hepatocellular carcinoma development that is associated with age-related changes in bile acid metabolism (Degirolamo et al. 2015). Obesity has also shown to decrease Fxr expression in adipose tissue (Cariou et al. 2006), and diabetes suppresses hepatic Fxr expression (Duran-Sandoval et al. 2004). In contrast, iFXR expression levels are not susceptible to obesity in mice and rats (Chen et al. 2010; Stenman et al. 2012).
3 Biology of Intestinal FXR
Understanding the physiological functions of iFXR allows prediction of the relevant metabolic processes that can be targeted by modulating its activity as well as of the potential side effects that could arise. Beneficial and adverse effects may both stem from either direct local FXR activity or from its transduction to other sites of the body by mediators such as hormones and growth factors or specific lipid species but also through adaptations in bile acid metabolism that lead to changes in bile acid pool composition, pool size, and/or cycling frequency.
3.1 iFXR, Intestinal Barrier Function, and Immunity
Composition of the microbiome, intestinal immune function, and integrity of the intestinal barrier are the major lines of defense at the intestinal surface, the largest surface of our body that is exposed to the outside world, and are all regulated by iFXR. Gut dysbiosis includes all three aspects and is a key feature of obesity and associated metabolic dysfunction, although we are only beginning to understand the mechanisms underlying these complex relationships (Teixeira et al. 2012 ; Sun et al. 2018b). It is well known that bile acids exert direct antibacterial effects in the small intestine, likely one of the reasons why bacterial numbers are low in the upper GI tract where bile acids enter (Binder et al. 1975; Ding et al. 1993). Vice versa, the microbiome also interacts with bile acid metabolism and iFXR signaling. Feeding the antioxidant tempol to mice inhibits iFXR signaling by altering the microbiome leading to an accumulation of intestinal tauro-β-muricholic acid (T-β-MCA), a potent FXR antagonist (Li et al. 2013). Importantly, iFXR was found to mediate the anti-obesity effects of tempol in high-fat diet-fed mice. Likewise, it was recently postulated that metformin, the first-line medication in the treatment of diabetes, acts in part by reducing numbers of Bacteroides fragilis, a bacterial species with bile acid-deconjugating activity, leading particularly to increased abundance of the FXR antagonist glycoursodeoxycholic acid (GUDCA) in humans. On the basis of an additional series of mouse experiments, showing that high-fat diet-fed mice colonized with Bacteroides fragilis showed more severe glucose intolerance and less sensitivity to the benefits of metformin treatment, it was concluded that metformin acts at least in part through the B. fragilis-GUDCA-iFXR axis to improve metabolic health (Sun et al. 2018a). Administration of the FXR agonist GW4064 abrogated small intestinal bacterial overgrowth and decreased bile duct ligation-induced intestinal permeability and inflammation in mice (Inagaki et al. 2006; Verbeke et al. 2015). Fxr-deficient mice are much more susceptible to these pathologies, i.e., tenfold higher aerobic bacterial counts were observed in lymph nodes in bile duct-ligated Fxr-deficient mice and even with a sham operation the lymph nodes of these mice already contained more aerobic bacteria than those of control animals. In accordance, the epithelial barrier of Fxr-deficient mice appeared to be deteriorated (Inagaki et al. 2006). Positive effects of iFXR activation are possibly in part mediated by the FXR targets inducible nitric oxide synthase (iNOS) and IL18 (Verbeke et al. 2015). A genetic variant in human FXR is associated with inflammatory bowel disease (Attinkara et al. 2012), and FXR activity in the ileum appears to be decreased in patients with Crohn’s disease (Nijmeijer et al. 2011). In a chemically induced colitis mouse model, the inflammatory response could be suppressed by the FXR agonist OCA in a Fxr-dependent manner (Vavassori et al. 2009). Furthermore, FXR agonism was shown to suppress expression of Toll-like receptor 4 (TLR4) and NF-κB regulated pro-inflammatory genes in colon macrophages, as also observed in liver (Vavassori et al. 2009; Wang et al. 2008). Similar outcomes of FXR agonism have been observed in a mouse model of intestinal ischemia and could therefore be relevant for pathologies in which aberrant gut-liver axis signaling plays a role (van Erpecum and Schaap 2015; Ceulemans et al. 2017). Indeed, the anti-inflammatory actions of FXR extend well beyond the intestine, for instance, also in blood leukocytes (Gadaleta et al. 2011a). Moreover, OCA treatment has similar effects in lipopolysaccharide-induced lung and kidney injury in mice, suggesting a general mechanism of action (Gai et al. 2016; Fei et al. 2019). Indeed, direct interaction of FXR with NF-kB signaling has been identified in many cell types as the underlying mechanism for NF-kB transrepression. This interaction may also explain reported bidirectional effects, as inflammation can in turn inhibit FXR activity, one of many ways by which metabolism and inflammation are intertwined (Gadaleta et al. 2011b; Verbeke et al. 2016). For instance, activation of membrane-bound TLR4 downregulates FXR expression in human monocytes, whereas intracellular TLRs such as TLR9 upregulate Fxr expression in human monocytes and in enterocytes of mice with colitis (Renga et al. 2013).
3.2 Modulation of Intestinal Cholesterol Metabolism by iFXR
The intestine plays an important role in the maintenance of cholesterol homeostasis by mediating its absorption from bile and diet as well as by active export, a process referred to as transintestinal cholesterol export (TICE, Temel and Brown 2015; de Boer et al. 2018). The majority of (dietary and biliary) cholesterol is absorbed proximally in the small intestine, and the presence of bile acids is an absolute requirement for absorption to occur (Voshol et al. 2001). The hydrophobicity of the intestinal bile acids impacts on lipid absorption in general, including cholesterol, due to the greater potency of hydrophobic bile acid species to incorporate lipid-soluble nutrients within the mixed micelles that are required for transport. Whether or not differences in bile acid pool composition contribute to the large variability in fractional cholesterol absorption between human subjects has remained unresolved so far (Bosner et al. 1999).
Besides absorption, the intestine can also regulate cholesterol turnover via TICE, a process that is subject to different modes of control. This route accounts for about 30% of fecal cholesterol loss in chow-fed mice as well as in humans (recently reviewed by de Boer et al. 2018). Inhibition of cholesterol absorption through inhibition of the cholesterol import transporter NPC1L1 by ezetimibe also increases the TICE pathway (Nakano et al. 2016; Jakulj et al. 2016), conceivably by preventing reuptake of cholesterol that is effluxed into the intestinal lumen by ABCG5/8 in enterocytes, leading to an increased net removal of cholesterol (de Boer et al. 2018). Interestingly, stimulation of FXR by the nonsteroidal agonist PX20606 (Abel et al. 2010 ) induced a strong increase of cholesterol removal via the TICE pathway in mice that was dependent on iFxr, i.e., an effect that was maintained in mice expressing Fxr only in the intestine (de Boer et al. 2017). Moreover, the effect on fecal cholesterol loss was completely additive to that of ezetimibe, causing extremely augmented cholesterol turnover upon combined treatment. Mechanistic experiments indicated a critical role for bile acid pool composition in the stimulation of TICE. In mice, iFXR activation and FGF19 administration were shown to shift the balance of the bile acid pool toward hydrophilic bile acid species, i.e., the muricholic acids, due to a relatively strong repression of Cyp8b1 expression (de Boer et al. 2017). These hydrophilic bile acid species may stimulate the activity of ABCG5/8 and hence cholesterol export into the lumen (Berge et al. 2000; Bonamassa and Moschetta 2013; de Boer et al. 2017). However, additional iFXR-dependent mechanisms may be operational as well, since fecal cholesterol excretion was only modestly increased in high-fat diet-fed Cyp8b1-deficient mice that show a similar hydrophilic bile acid pool composition (Bonde et al. 2016). Whether or not therapeutic targeting of iFXR may be beneficial for treatment of hypercholesterolemia to prevent cardiovascular disease in humans remains to be established. Stimulation of TICE via modulation of bile acid pool composition is not expected to be conserved in humans based on the marked differences in pool composition between humans and mice. In fact, in humans CYP8B1 repression will theoretically lead to a CDCA-dominated, hydrophobic bile acid pool and hence more effective cholesterol absorption and less TICE.
An additional layer of complexity in the role of iFXR in control of cholesterol absorption was recently described by Kim et al. (2018), showing that iFXR-mediated release of Fgf15 from the murine ileum signals to the proximal small intestine to repress expression of Npc1l1 and, thereby, cholesterol absorption. In a series of experiments, exploiting Shp-deficient and Fgf15-deficient mice, data were presented to indicate that ileum-derived Fgf15 as well as i.v. injected FGF19 led to phosphorylation of SHP which suppressed activity of sterol regulatory transcription factor 2 (SREBPF2) that regulates sterol-activated transcription of Npc1l1 in enterocytes of the upper small intestine. Thus, FGF15/19 may not only modulate the cholesterol-solubilizing capacity of mixed micelles in the lumen of the small intestine but also the expression of the cholesterol transporter at the apical membrane of the enterocytes. Consequently, the overall effect of iFXR activation and Fgf15 release in mice is a strong acceleration of cholesterol turnover by stimulation of TICE and reduction of fractional cholesterol absorption, the latter by a dual mode of action. Whether this also applies to humans, with a more hydrophobic bile acid pool, remains to be established. At the same time, obviously, suppression of hepatic bile acid synthesis, which comprises a very important pathway for cholesterol turnover in humans as well as rodents, will impair (hepatic) cholesterol turnover. This may underlie (part of) the reported LDL elevations upon pharmacological FXR activation (Pencek et al. 2016; Neuschwander-Tetri et al. 2015).
3.3 Role of iFXR in Control of Glucose Metabolism
In the past decade, several studies have reported a role for FXR in control of glucose metabolism, in most cases employing whole-body Fxr knockout mouse models and/or systemically acting FXR modulators. Yet, some interesting features concerning specific roles of iFXR in control of intestinal and whole-body glucose metabolism with potential therapeutic potential have also been reported. To the best of our knowledge, only a single report so far has demonstrated a role for iFXR, particularly in the proximal small intestine, in the kinetics of glucose absorption. Employing different stable isotopically labeled glucose tracers and compartment modelling, van Dijk et al. (2009) demonstrated that absorption of glucose is delayed in whole-body Fxr-deficient mice, due to an increased flux of glucose molecules through the glucose-6-phosphate pool in enterocytes before entering the bloodstream. Thus, while uptake of glucose by enterocytes was similar in wild-type and Fxr-deficient mice, the residence time of glucose molecules within the enterocytes was longer in Fxr-deficient mice due to enhanced hexokinase 1 and 2 (Hk1/2)-mediated phosphorylation. Glucose-6-phosphate subsequently requires dephosphorylation by glucose-6-phosphatase before glucose can be released via this indirect pathway into the blood. Indeed, the expression of Hk1 and Hk2 was sixfold higher in the proximal part of the small intestine of Fxr-deficient mice compared to the controls while all other transporters and enzymes involved in glucose handling were unaffected. Whether or not the kinetics of intestinal glucose absorption is modulated upon pharmacological FXR activation remains to be established.
Effects of a deficiency in iFXR on systemic glucose metabolism have been studied in more detail. Fxr and Tgr5 are co-expressed in enteroendocrine L cells, and activation of TGR5 by luminal bile acids regulates intestinal production of the incretin glucagon-like peptide 1 (GLP-1) that in turn stimulates pancreatic insulin secretion and glucose homeostasis (Pathak et al. 2017; Thomas et al. 2009). GLP-1 and gastric inhibitory peptide (GIP) are the most important incretins and the basis of incretin mimetic therapies for type 2 diabetes, as well as dipeptidyl peptidase-4 (DPP-4) inhibitors that prevent the degradation of endogenous incretins (reviewed in Ahrén 2012). FXR appears to modulate Tgr5 expression, stimulating GLP-1, yet FXR also suppresses preproglucagon, encoding GLP-1 (Trabelsi et al. 2015). A dynamic interaction between FXR and TGR5 was subsequently proposed to potentiate GLP-1 action in the control of glycemic control (Kim and Fang 2018).
Low serum FGF19 levels were associated with high fasting plasma glucose levels and type 2 diabetes (Fang et al. 2013), and FGF19 action has been implicated in hepatic glucose metabolism (Potthoff et al. 2011). However, long-term central effects of FGF19 have also been shown to contribute to glucose lowering upon a single intracerebroventricular injection (Fu et al. 2004; Morton et al. 2013). Interestingly, a role for the central nervous system was recently confirmed by showing its dependence on β-Klotho specifically in the nervous system, while the acute glucose-lowering effects were shown to depend on β-Klotho in adipose tissue (Lan et al. 2017).
Paradoxically, there is recent evidence supporting that inhibition, rather than activation, of iFxr improves glucose metabolism during metabolic disease conditions. Mice with iFxr deficiency are protected from diet-induced obesity and insulin resistance (Li et al. 2013; Jiang et al. 2015a; Xie et al. 2017). In addition, obese iFxr-deficient mice treated with tempol, which reduces microbiota species with high BSH activity and therefore increases levels of the FXR antagonist T-β-MCA, lacked the observed decrease in blood glucose and insulin levels that occur in control animals, indicating the significance of iFXR for this approach (Li et al. 2013). These effects were associated with decreased ceramide levels and could be reversed by ceramide administration (Jiang et al. 2015b). The translational potential of these studies, however, remains uncertain, as humans do not produce muricholic acids. To overcome this translational issue, mice with a humanized bile acid pool have recently been generated by deleting the Cyp2C cluster (Takahashi et al. 2016).
3.4 iFXR and Energy Metabolism
The potential of iFXR to control energy homeostasis has attracted strong interest in the development of therapeutics in the management of various metabolic syndrome-associated morbidities. FGF15/19 is an important mediator of these effects. Transgenic mice constitutively expressing FGF19 in muscle have increased brown adipose tissue size and energy expenditure resulting in reduced body weight upon high-fat diet feeding. FGF19 treatment of obese mice has similar effects (Tomlinson et al. 2002; Fu et al. 2004). Despite a low sequence similarity between murine Fgf15 and human FGF19, both genes are syntenic, and their biological function in the regulation of bile acid homeostasis is well conserved. As FGF19 protein has a stability superior to Fgf15, transgenic expression and treatments have almost exclusively been performed with FGF19. The interaction between iFXR and the microbiome is also one of the major routes by which microbiota impact host metabolism and therefore represents an indirect approach to target iFXR (Sayin et al. 2013; Degirolamo et al. 2014; Sun et al. 2017). Besides beneficial metabolic effects, it was also shown that microbiota-induced obesity in mice requires iFxr (Li et al. 2013; Zhang et al. 2016; Parséus et al. 2017).
Next to suppression of bile acid synthesis, many other hepatic processes are under iFXR control as well, depending for a large part on FGF15/19 signaling.
3.5 Hepatic Metabolism and iFXR
Hepatic activation of FXR exerts beneficial effects in nonalcoholic fatty liver disease (NAFLD) by repressing de novo lipogenesis and promoting fatty acid oxidation and VLDL clearance (Zhang and Edwards 2008). Clinical trials with OCA have shown initial improvements in histological features of NASH (Neuschwander-Tetri et al. 2015), although more studies are needed to assess long-term efficacy and safety. Release of FGF15/19 by iFXR activation and subsequent binding to fibroblast growth factor receptor 4 (FGFR4) and its co-receptor β-Klotho on hepatocytes initiates a signaling cascade that not only suppresses bile acid synthesis but also gluconeogenesis and promotes protein synthesis (Kir et al. 2011). Furthermore, FGF15/19 decreases hepatic lipogenesis (Fuchs et al. 2016) and could indirectly stimulate mitochondrial fatty acid oxidation (Tomlinson et al. 2002; Fu et al. 2004). Despite these premises, there is also evidence supporting that iFXR inhibition rather than activation could improve NAFLD. Mice lacking iFXR expression remain lean during a high-fat diet challenge, possibly as a consequence of lower intestinal production of ceramides, resulting in the downregulation of Srebp1c and thus decreased lipogenesis (Jiang et al. 2015a). Similar results were obtained with pharmacological ASBT inhibition that reduces iFXR ligand availability and paradoxically also with inhibition of iFXR (described below). A successful phase II clinical trial was recently reported in NASH patients with NGM282 an FGF19 analogue (Harrison et al. 2018). FGF19 administration in mice also represses hepatic gluconeogenesis via cAMP response element-binding protein (CREB) (Potthoff et al. 2011). These effects of FGF19 seem analogous to the actions of insulin; however, they occur independently and through different pathways (Kir et al. 2011).
3.6 Temporal Regulation of iFXR and Oncogenic Effects
Besides studies on iFXR that are based on tissue-specific receptor deficiency, tissue-specific constitutively active overexpression has also been applied, albeit mostly to evaluate effects on tumorigenesis. These studies showed that transgenic mice with an intestine-specific constitutive expression of Fxr (iVP16FXR) were protected from hepatocellular carcinoma (Degirolamo et al. 2015). Intestinal FXR targets such as Fgf15, Shp, and Ostα/β were affected in iVP16FXR mice, and a 30% reduction in bile acid pool was observed, as well as both decreased inflammation and tissue damage in intestine and in liver in models of cholestasis (Modica et al. 2012). Metabolic effects from this constitutively active model are however difficult to translate, as endogenous FXR activity oscillates and occurs predominantly postprandially. We and others identified iFXR as a modulator of hepatic diurnal rhythm through FXR-FGF15 signaling (Stroeve et al. 2010), and plasma Fgf15 also showed an FXR-dependent circadian rhythm (Katafuchi et al. 2015). In line with a physiological function in the postprandial phase is the observation of suppressive actions of FXR on the intracellular quality control mechanism, autophagy, that is mostly active in the fasted state (Lee et al. 2014; Seok et al. 2014). As autophagy is a pathway that degrades superfluous components in the cell, both inactive and overly active autophagies are detrimental. Therefore, autophagy is highly interlinked with the circadian clock, and strict temporal regulation is warranted (Toledo et al. 2018). FGF19 also transduces a suppressive signal on autophagy in the liver (Byun et al. 2017). Moreover, regulation by FXR of Sqstm1 encoding P62, an important factor in selective autophagy, occurred especially in the intestine (Williams et al. 2012). Likewise, the second proteolytic quality control mechanism, the unfolded protein response (UPR) that is known to respond to a meal (Liu et al. 2018), also has a cytoprotective function if kept at bay. Sustained activation of the UPR, however, for example, during chronic ER stress, can lead to disease (Fu et al. 2012) and aging may downregulate hepatic FXR due to sustained ER stress, leading to hepatic steatosis (Xiong et al. 2014). For instance, constitutive activity of ATF6, one of the three UPR sensors in the intestine, leads to loss of intestinal barrier function and microbiota-dependent tumorigenesis (Coleman et al. 2018). Albeit less established, recent findings have also identified hepatic FXR as a regulator of the other two UPR sensors PERK and IRE1α/XBP1 (Liu et al. 2018; Han et al. 2018). These interactions suggest that a tight temporal regulation of iFXR is required to maintain cellular homeostasis, as both over- and underrepresentation of iFXR output has been implicated in tumor development.
3.6.1 Oncogenic Potential of iFXR Modulation
As FGF15/19 is an endocrine member of the FGF family that has been implicated in the control of cellular proliferation and development, modulating iFXR may come with some potential carcinogenic risks. Indeed, Fgf15 overexpression induces hepatocyte proliferation in mice, independently of hepatic bile acid levels, both with and without previous partial hepatectomy (Kong et al. 2012 ). FGF19 transgenic mice develop hepatocellular carcinomas that is dependent of FGFR4 (Nicholes et al. 2002; French et al. 2012), and as a result FGF19 has been coined an oncogene (Cui et al. 2018). Interestingly, FGF19 but not Fgf15 induces hepatocellular carcinoma at supraphysiological levels in mouse models of metabolic syndrome, perhaps due to the low stability of the latter (Zhou et al. 2017). In humans, increased FGF19 expression was found in hepatocellular carcinoma and correlated with tumor progression (Miura et al. 2012). In contrast, in mouse colon, Fgf15 deficiency resulted in increased cellular proliferation, crypt-villus length, and advanced neoplasia (Cheng et al. 2018). Similar effects were also observed in Asbt-deficient mice, possibly due to increased TGR5 signaling (Thulesen 2004; Raufman et al. 2015). Additionally, Fxr deficiency promotes intestinal tumor development in different models of colon cancer, by increasing cell proliferation and inflammation (Modica et al. 2008; Maran et al. 2009). In line with a metabolic rheostat function for FXR, potential carcinogenic risks exist upon unbalanced activation as well as deactivation that must be taken into account when designing drugs that target iFXR.
4 Approaches for Intestine-Specific Targeting of FXR
As interference with bile acid-responsive receptors can invoke potent physiological responses, generation of novel modulators with selectivity for a single receptor or even a subset of its target genes has gained substantial interest (Massafra et al. 2018). Studies reporting pharmacological approaches to either activate or inhibit iFXR and the physiological consequences will be discussed below.
4.1 Selective Activation of iFXR: Fexaramine
The observation that the FXR agonist fexaramine (Fex) is poorly absorbed by the intestine but still mediates potent activation of iFXR opened up an avenue to specifically target iFXR (Fang et al. 2015). Obese mice treated for 5 weeks showed reduced fat mass and metabolic improvement as well as reduced systemic inflammation. These beneficial effects were attributed to increased energy expenditure due to brown adipose tissue activity, browning of white adipose tissue, and a shift in bile acid composition favorable for TGR5 activity. Indeed, in line with a role for TGR5 in activating brown adipose tissue (Watanabe et al. 2006), the effects were partially dependent on whole-body Tgr5 (Fang et al. 2015). These findings were recently confirmed in obese leptin receptor-deficient db/db mice and extended by showing a change in microbiota composition upon Fex treatment and reversal of the Fex-induced metabolic improvement after antibiotic treatment (Pathak et al. 2018).
4.2 Selective Inhibition of iFXR
Similar to the situation in obese mice treated with tempol, which reduces microbial conversion of the FXR inhibitor T-β-MCA, treatment of mice with a microbial BSH-resistant synthetic iFXR inhibitor, glycine-β-MCA, also reduced blood glucose and insulin levels (Jiang et al. 2015b). These effects were associated with reduced systemic ceramide levels and could be reversed by ceramide administration. Similar beneficial results were obtained by administering caffeic acid phenethyl ester (CAPE), which inhibits BSH and increases T-β-MCA levels. (Xie et al. 2017). Despite the fact that the translational relevance of these studies is limited, since humans do not produce muricholic acids, this work substantiates the claim that modulation of microbial species that control bile acid metabolism appears to be a viable approach for targeting host metabolism.
4.3 Indirect Pharmacological Approaches That Target iFXR
Another viable approach to modulate iFXR activity is through manipulation of intestinal bile acid transporters. In this scenario, Asbt deficiency or Abst inhibition will have an antagonizing effect, whereas Ostα/ Ostβ deficiency or blockade will result in activation of iFXR. Indeed, Asbt-deficient mice show increased fecal bile acid excretion, decreased ileal Fgf15 expression, increased hepatic Cyp7a1 expression, and a bile acid pool predominantly consisting of cholic acid. In contrast, Ostα-deficient mice show increased ileal Fgf15 expression, suppressed hepatic Cyp7a1 expression (reviewed in Dawson 2017), and decreased lipid absorption accompanied by modestly improved insulin sensitivity (Wheeler et al. 2014). Despite the fact that Ostα-deficient mice suffer from bile acid-induced hepatic injury in the postnatal period (Ferrebee et al. 2018), approaches to stimulate iFXR by pharmacological inhibition of OSTα/OSTβ have recently been investigated, supporting a role for this transporter as a drug target and its inhibitor clofazimine as a modulator of iFXR (van de Wiel et al. 2018 ).
Remarkably, inhibition of ASBT, which prevents iFXR activation, has also been shown to be beneficial for glycemic control (Chen et al. 2012; Wu et al. 2013; Rao et al. 2016), similar to bile acid sequestrants, which decrease iFXR signaling as a consequence of reduced bile acid reabsorption (Handelsman 2011). Pharmacological interventions such as bile acid-binding resins and ASBT or OSTα-β inhibitors increase fecal bile acid loss by preventing their reuptake by enterocytes or their subsequent release into portal blood. Obviously, potential adverse effects of an increased exposure of the colon to bile acids need to be taken into account during evaluation of these approaches. Increased fecal loss of bile acids will be compensated for by increased hepatic bile acid synthesis from cholesterol, thus inducing cholesterol synthesis and hepatic LDL receptor expression and accelerating LDL uptake from the blood compartment (Slijepcevic and van de Graaf 2017). ASBT inhibition was shown to reduce hepatic lipid accumulation in high-fat diet-fed mice (Rao et al. 2016), although this beneficial effect could also result from fat malabsorption due to a decreased bile acid pool.
5 Perspective
Beyond its well-known functions in regulating bile acid homeostasis and ensuring cellular protection from bile acid toxicity, intestinal FXR is clearly also involved in the regulation of various aspects of energy metabolism, cellular proliferation, and intestinal barrier function, among others. These pleiotropic effects favor intestinal FXR as a potentially attractive target for a variety of diseases but, at the same time, also provide a basis for relevant adverse effects. In particular, the mitogenic risk of intestinal FXR modulation is of concern. More studies are needed to reconcile discordant findings surrounding intestinal FXR targeting in energy metabolism and to define the role of FXR in each relevant disease state. The translational value of murine studies must be verified for each pathway and disease under evaluation. The generation of mouse models with a “humanized” bile acid pool will constitute a valuable first step in clarifying some of these issues (Takahashi et al. 2016). Particularly, the consequences of iFXR modulation on intestinal carbohydrate metabolism require additional studies since, in our opinion, this aspect of iFXR biology has not sufficiently been addressed so far. Also, the potential consequences of posttranscriptional modulation of iFXR have not been sufficiently addressed to date. Importantly, as humans have highly varying inter-individual bile acid and microbiota profiles, “personalized medicine” approaches are likely to be relevant for effective intestinal FXR modulation. Notably, the only treatment with proven therapeutic benefits in humans so far, both with respect to hypercholesterolemia and glucose metabolism, is based on sequestration of bile acids, i.e., reduction of iFXR activation. Last but not least, the long-term effects of intestinal FXR agonism and/or antagonism need to be assessed, since for both beneficial effects have been reported in mouse studies, particularly with respect to the potential consequences of manipulating circadian rhythms of bile acid signaling.
References
Abdelkarim M, Caron S, Duhem C et al (2010) The farnesoid X receptor regulates adipocyte differentiation and function by promoting peroxisome proliferator-activated receptor-gamma and interfering with the Wnt/beta-catenin pathways. J Biol Chem 285:36759–36767
Abel U, Schlüter T, Schulz A et al (2010) Synthesis and pharmacological validation of a novel series of non-steroidal FXR agonists. Bioorg Med Chem Lett 20:4911–4917
Ahrén B (2012) DPP-4 inhibition and islet function. J Diabetes Investig 3:3–10
Albaugh VL, Banan B, Antoun J et al (2018) Role of bile acids and GLP-1 in mediating the metabolic improvements of bariatric surgery. Gastroenterology 156:1041–1051.e4
Armstrong LE, Guo GL (2017) Role of FXR in liver inflammation during nonalcoholic steatohepatitis. Curr Pharmacol Rep 3:92–100
Attinkara R, Mwinyi J, Truninger K et al (2012) Association of genetic variation in the NR1H4 gene, encoding the nuclear bile acid receptor FXR, with inflammatory bowel disease. BMC Res Notes 5:461
Balasubramaniyan N, Ananthanarayanan M, Suchy FJ (2012) Direct methylation of FXR by Set7/9, a lysine methyltransferase, regulates the expression of FXR target genes. Am J Physiol Gastrointest Liver Physiol 302:G937–G947
Balasubramaniyan N, Luo Y, Sun AQ et al (2013) SUMOylation of the farnesoid X receptor (FXR) regulates the expression of FXR target genes. J Biol Chem 288:13850–13862
Becares N, Gage MC, Pineda-Torra I (2017) Posttranslational modifications of lipid-activated nuclear receptors: focus on metabolism. Endocrinology 158:213–225
Benhamed F, Filhoulaud G, Caron S et al (2015) O-GlcNAcylation links ChREBP and FXR to glucose-sensing. Front Endocrinol 5:230
Berge KE, Tian H, Graf GA et al (2000) Accumulation of dietary cholesterol in sitosterolemia caused by mutations in adjacent ABC transporters. Science 290:1771–1775
Berrabah W, Aumercier P, Gheeraert C et al (2014) Glucose sensing O-GlcNAcylation pathway regulates the nuclear bile acid receptor farnesoid X receptor (FXR). Hepatology 59:2022–2033
Bertolini A, van de Peppel IP, Doktorova-Demmin M et al (2019) Defective FXR-FGF15 signaling and bile acid homeostasis in cystic fibrosis mice can be restored by the laxative polyethylene glycol. Am J Physiol Gastrointest Liver Physiol 316:G404–G411
Bilodeau S, Caron V, Gagnon J et al (2017) A CK2-RNF4 interplay coordinates non-canonical SUMOylation and degradation of nuclear receptor FXR. J Mol Cell Biol 9:195–208
Binder HJ, Filburn B, Floch M (1975) Bile acid inhibition of intestinal anaerobic organisms. Am J Clin Nutr 28:119–125
Boesjes M, Bloks VW, Hageman J et al (2014) Hepatic farnesoid X-receptor isoforms α2 and α4 differentially modulate bile salt and lipoprotein metabolism in mice. PLoS One 9:e115028
Bonamassa B, Moschetta A (2013) Atherosclerosis: lessons from LXR and the intestine. Trends Endocrinol Metab 24:120–128
Bonde Y, Eggertsen G, Rudling M (2016) Mice abundant in muricholic bile acids show resistance to dietary induced steatosis, weight gain, and to impaired glucose metabolism. PLoS One 11:e0147772
Bosner MS, Lange LG, Stenson WF (1999) Percent cholesterol absorption in normal women and men quantified with dual stable isotopic tracers and negative ion mass spectrometry. J Lipid Res 40:302–308
Brufau G, Stellaard F, Prado K et al (2010) Improved glycemic control with colesevelam treatment in patients with type 2 diabetes is not directly associated with changes in bile acid metabolism. Hepatology 52:1455–1464
Bull LN, van Eijk MJ, Pawlikowska L et al (1998) A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis. Nat Genet 18:219–224
Byun S, Kim YC, Zhang Y et al (2017) A postprandial FGF19-SHP-LSD1 regulatory axis mediates epigenetic repression of hepatic autophagy. EMBO J 36:1755–1769
Cariello M, Piccinin E, Garcia-Irigoyen O et al (2018) Nuclear receptor FXR, bile acids and liver damage: introducing the progressive familial intrahepatic cholestasis with FXR mutations. Biochim Biophys Acta Mol basis Dis 1864:1308–1318
Cariou B, van Harmelen K, Duran-Sandoval D et al (2006) The farnesoid X receptor modulates adiposity and peripheral insulin sensitivity in mice. J Biol Chem 281:11039–11049
Ceulemans LJ, Verbeke L, Decuypere J-P et al (2017) Farnesoid X receptor activation attenuates intestinal ischemia reperfusion injury in rats. PLoS One 12:e0169331
Charach G, Argov O, Geiger K et al (2017) Diminished bile acids excretion is a risk factor for coronary artery disease: 20-year follow up and long-term outcome. Ther Adv Gastroenterol 4:1756283–7743420
Chen L, McNulty J, Anderson D et al (2010) Cholestyramine reverses hyperglycemia and enhances glucose-stimulated glucagon-like peptide 1 release in Zucker diabetic fatty rats. J Pharmacol Exp Ther 334:164–170
Chen L, Yao X, Young A et al (2012) Inhibition of apical sodium-dependent bile acid transporter as a novel treatment for diabetes. Am J Physiol Endocrinol Metab 302:68–76
Chen WG, Zheng JX, Xu X et al (2018) Hippocampal FXR plays a role in the pathogenesis of depression: a preliminary study based on lentiviral gene modulation. Psychiatry Res 264:374–379
Cheng K, Metry M, Felton J et al (2018) Diminished gallbladder filling, increased fecal bile acids, and promotion of colon epithelial cell proliferation and neoplasia in fibroblast growth factor 15-deficient mice. Oncotarget 9:25572–25585
Choi M, Moschetta A, Bookout AL et al (2006) Identification of a hormonal basis for gallbladder filling. Nat Med 12:1253–1255
Coleman OI, Lobner EM, Bierwirth S et al (2018) Activated ATF6 induces intestinal dysbiosis and innate immune response to promote colorectal tumorigenesis. Gastroenterology 155:1539–1552
Cui G, Martin RC, Jin H et al (2018) Up-regulation of FGF15/19 signaling promotes hepatocellular carcinoma in the background of fatty liver. J Exp Clin Cancer Res 37:136
Dawson PA (2017) Roles of Ileal ASBT and OSTα-OSTβ in regulating bile acid signaling. Dig Dis 35:261–266
de Boer JF, Schonewille M, Boesjes M et al (2017) Intestinal farnesoid X receptor controls transintestinal cholesterol excretion in mice. Gastroenterology 152:1126–1138
de Boer JF, Kuipers F, Groen AK (2018) Cholesterol transport revisited: a new turbo mechanism to drive cholesterol excretion. Trends Endocrinol Metab 29:123–133
Degirolamo C, Rainaldi S, Bovenga F et al (2014) Microbiota modification with probiotics induces hepatic bile acid synthesis via downregulation of the Fxr-Fgf15 axis in mice. Cell Rep 7:12–18
Degirolamo C, Modica S, Vacca M et al (2015) Prevention of spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice by intestinal-specific farnesoid X receptor reactivation. Hepatology 61:161–170
Ding JW, Andersson R, Soltesz V et al (1993) The role of bile and bile acids in bacterial translocation in obstructive jaundice in rats. Eur Surg Res 25:11–19
Duran-Sandoval D, Mautino G, Martin G et al (2004) Glucose regulates the expression of the farnesoid X receptor in liver. Diabetes 53:890–898
Fang Q, Li H, Song Q et al (2013) Serum fibroblast growth factor 19 levels are decreased in Chinese subjects with impaired fasting glucose and inversely associated with fasting plasma glucose levels. Diabetes Care 36:2810–2814
Fang S, Suh JM, Reilly SM et al (2015) Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat Med 21:159–165
Fedorowski T, Salen G, Tint GS et al (1979) Transformation of chenodeoxycholic acid and ursodeoxycholic acid by human intestinal bacteria. Gastroenterology 77:1068–1073
Fei J, Fu L, Hu B et al (2019) Obeticholic acid alleviate lipopolysaccharide-induced acute lung injury via its anti-inflammatory effects in mice. Int Immunopharmacol 66:177–184
Ferrebee CB, Li J, Haywood J et al (2018) Organic solute transporter α-β protects ileal enterocytes from bile acid-induced injury. Cell Mol Gastroenterol Hepatol 5:499–522
Frankenberg T, Miloh T, Chen FY et al (2008) The membrane protein ATPase class I type 8B member 1 signals through protein kinase C zeta to activate the farnesoid X receptor. Hepatology 48:1896–1905
French DM, Lin BC, Wang M et al (2012) Targeting FGFR4 inhibits hepatocellular carcinoma in preclinical mouse models. PLoS One 7:e36713
Friedman ES, Li Y, Shen TD et al (2018) FXR-dependent modulation of the human small intestinal microbiome by the bile acid derivative obeticholic acid. Gastroenterology 155:1741–1752
Fu L, John LM, Adams SH et al (2004) Fibroblast growth factor 19 increases metabolic rate and reverses dietary and leptin-deficient diabetes. Endocrinology 145:2594–2603
Fu S, Watkins SM, Hotamisligil GS (2012) The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab 15:623–634
Fuchs CD, Traussnigg SA, Trauner M (2016) Nuclear receptor modulation for the treatment of nonalcoholic fatty liver disease. Semin Liver Dis 36:69–86
Gadaleta RM, van Erpecum KJ, Oldenburg B et al (2011a) Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut 60:463–472
Gadaleta RM, Oldenburg B, Willemsen ECL et al (2011b) Activation of bile salt nuclear receptor FXR is repressed by pro-inflammatory cytokines activating NF-κB signaling in the intestine. Biochim Biophys Acta Mol basis Dis 1812:851–858
Gai Z, Gui T, Hiller C et al (2016) Farnesoid X receptor protects against kidney injury in uninephrectomized obese mice. J Biol Chem 291:2397–2411
Gineste G, Sirvent A, Paumelle R et al (2008) Phosphorylation of farnesoid X receptor by protein kinase C promotes its transcriptional activity. Mol Endocrinol 22:2433–2447
Goodwin B, Jones SA, Price RR et al (2000) A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell 6:517–526
Gray MA, Squires EJ (2015) Investigation of the dominant positive effect of porcine farnesoid X receptor (FXR) splice variant 1. Gene 560:71–76
Han CY, Rho HS, Kim A et al (2018) FXR inhibits endoplasmic reticulum stress-induced NLRP3 inflammasome in hepatocytes and ameliorates liver injury. Cell Rep 24:2985–2999
Handelsman Y (2011) Role of bile acid sequestrants in the treatment of type 2 diabetes. Diabetes Care 34:S244–S250
Harrison SA, Rinella ME, Abdelmalek MF et al (2018) NGM282 for treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 391:1174–1185
Herman-Edelstein M, Weinstein T, Levi M (2018) Bile acid receptors and the kidney. Curr Opin Nephrol Hypertens 27:56–62
Heuman DM (1989) Quantitative estimation of the hydrophilic-hydrophobic balance of mixed bile salt solutions. J Lipid Res 30:719–730
Hill CJ, Lynch DB, Murphy K et al (2017) Evolution of gut microbiota composition from birth to 24 weeks in the INFANTMET cohort. Microbiome 5:4
Hofmann AF, Hagey LR (2014) Key discoveries in bile acid chemistry and biology and their clinical applications: history of the last eight decades. J Lipid Res 55:1553–1595
Huber RM, Murphy K, Miao B et al (2002) Generation of multiple farnesoid-X-receptor isoforms through the use of alternative promoters. Gene 290:35–43
Inagaki T, Choi M, Moschetta A et al (2005) Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab 2:217–225
Inagaki T, Moschetta A, Lee Y-K et al (2006) Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci 103:3920–3925
Jakulj L, van Dijk TH, de Boer JF et al (2016) Transintestinal cholesterol transport is active in mice and humans and controls ezetimibe-induced fecal neutral sterol excretion. Cell Metab 24:783–794
Jiang C, Xie C, Li F et al (2015a) Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J Clin Invest 125:386–402
Jiang C, Xie C, Lv Y et al (2015b) Intestine-selective farnesoid X receptor inhibition improves obesity-related metabolic dysfunction. Nat Commun 6:10166
Joyce SA, Gahan CGM (2016) Bile acid modifications at the microbe-host interface: potential for nutraceutical and pharmaceutical interventions in host health. Annu Rev Food Sci Technol 7:313–333
Katafuchi T, Esterházy D, Lemoff A et al (2015) Detection of FGF15 in plasma by stable isotope standards and capture by anti-peptide antibodies and targeted mass spectrometry. Cell Metab 21:898–904
Kemper JK, Xiao Z, Ponugoti B et al (2009) FXR acetylation is normally dynamically regulated by p300 and SIRT1 but constitutively elevated in metabolic disease states. Cell Metab 10:392–404
Kim H, Fang S (2018) Crosstalk between FXR and TGR5 controls glucagon-like peptide 1 secretion to maintain glycemic homeostasis. Lab Anim Res 34:140–146
Kim I, Ahn SH, Inagaki T et al (2007) Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J Lipid Res 48:2664–2672
Kim DH, Xiao Z, Kwon S et al (2014) A dysregulated acetyl/SUMO switch of FXR promotes hepatic inflammation in obesity. EMBO J 34:184–199
Kim KH, Choi S, Zhou Y et al (2017) Hepatic FXR/SHP axis modulates systemic glucose and fatty acid homeostasis in aged mice. Hepatology 66:498–509
Kim Y-C, Byun S, Seok S et al (2018) Small heterodimer partner and fibroblast growth factor 19 inhibit expression of NPC1L1 in mouse intestine and cholesterol absorption. Gastroenterology 156:1052–1065
Kir S, Beddow SA, Samuel VT et al (2011) FGF19 as a postprandial, insulin-independent activator of hepatic protein and glycogen synthesis. Science 331:1621–1624
Kong B, Wang L, Chiang JYL et al (2012) Mechanism of tissue-specific farnesoid X receptor in suppressing the expression of genes in bile-acid synthesis in mice. Hepatology 56:1034–1043
Kuipers F, Bloks VW, Groen AK (2014) Beyond intestinal soap – bile acids in metabolic control. Nat Rev Endocrinol 10:488–498
Lan T, Morgan DA, Rahmouni K et al (2017) FGF19, FGF21, and an FGFR1/β-Klotho-activating antibody act on the nervous system to regulate body weight and glycemia. Cell Metab 26:709–718
Lee J, Kemper JK (2010) Controlling SIRT1 expression by microRNAs in health and metabolic disease. Aging 2:527–534
Lee J, Seok S, Yu P et al (2012) Genomic analysis of hepatic farnesoid X receptor binding sites reveals altered binding in obesity and direct gene repression by farnesoid X receptor in mice. Hepatology 56:108–117
Lee JM, Wagner M, Xiao R et al (2014) Nutrient-sensing nuclear receptors coordinate autophagy. Nature 516:112–115
Lefebvre P, Cariou B, Lien F et al (2009) Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev 89:147–191
Li F, Jiang C, Krausz KW et al (2013) Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity. Nat Commun 4:2384
Lien F, Berthier A, Bouchaert E et al (2014) Metformin interferes with bile acid homeostasis through AMPK-FXR crosstalk. J Clin Invest 124:1037–1051
Liu X, Guo GL, Kong B et al (2018) Farnesoid X receptor signaling activates the hepatic X-box binding protein 1 pathway in vitro and in mice. Hepatology 68:304–316
Lu TT, Makishima M, Repa JJ et al (2000) Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell 6:507–515
Luo L, Aubrecht J, Li D et al (2018) Assessment of serum bile acid profiles as biomarkers of liver injury and liver disease in humans. PLoS One 13:e0193824
Makishima M, Okamoto AY, Repa JJ et al (1999) Identification of a nuclear receptor for bile acids. Science 284:1362–1365
Maran RRM, Thomas A, Roth M et al (2009) Farnesoid X receptor deficiency in mice leads to increased intestinal epithelial cell proliferation and tumor development. J Pharmacol Exp Ther 328:469–477
Maruyama T, Miyamoto Y, Nakamura T et al (2002) Identification of membrane-type receptor for bile acids (M-BAR). Biochem Biophys Res Commun 298:714–719
Massafra V, Pellicciari R, Gioiello A et al (2018) Progress and challenges of selective farnesoid X receptor modulation. Pharmacol Ther 191:162–177
Miura S, Mitsuhashi N, Shimizu H et al (2012) Fibroblast growth factor 19 expression correlates with tumor progression and poorer prognosis of hepatocellular carcinoma. BMC Cancer 12:56
Modica S, Murzilli S, Salvatore L et al (2008) Nuclear bile acid receptor FXR protects against intestinal tumorigenesis. Cancer Res 68:9589–9594
Modica S, Petruzzelli M, Bellafante E et al (2012) Selective activation of nuclear bile acid receptor FXR in the intestine protects mice against cholestasis. Gastroenterology 142:355–365
Morton GJ, Matsen ME, Bracy DP et al (2013) FGF19 action in the brain induces insulin-independent glucose lowering. J Clin Invest 123:4799–4808
Mowat AM, Agace WW (2014) Regional specialization within the intestinal immune system. Nat Rev Immunol 14:667–685
Mueller M, Thorell A, Claudel T et al (2015) Ursodeoxycholic acid exerts farnesoid X receptor-antagonistic effects on bile acid and lipid metabolism in morbid obesity. J Hepatol 62:1398–1404
Nakano T, Inoue I, Takenaka Y et al (2016) Ezetimibe promotes brush border membrane-to-lumen cholesterol efflux in the small intestine. PLoS One 11:e0152207
Neuschwander-Tetri BA, Loomba R, Sanyal AJ et al (2015) Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet 385:956–965
Nevens F, Andreone P, Mazzella G et al (2016) A placebo-controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med 375:631–643
Nicholes K, Guillet S, Tomlinson E et al (2002) A mouse model of hepatocellular carcinoma: ectopic expression of fibroblast growth factor 19 in skeletal muscle of transgenic mice. Am J Pathol 160:2295–2307
Nijmeijer RM, Gadaleta RM, van Mil SWC et al (2011) Farnesoid X receptor (FXR) activation and FXR genetic variation in inflammatory bowel disease. PLoS One 6:e23745
Otte K, Kranz H, Kober I et al (2003) Identification of farnesoid X receptor beta as a novel mammalian nuclear receptor sensing lanosterol. Mol Cell Biol 23:864–872
Out C, Patankar JV, Doktorova M et al (2015) Gut microbiota inhibit Asbt-dependent intestinal bile acid reabsorption via Gata4. J Hepatol 63:697–704
Overington JP, Al-Lazikani B, Hopkins AL (2006) How many drug targets are there? Nat Rev Drug Discov 5:993–996
Parks DJ, Blanchard SG, Bledsoe RK et al (1999) Bile acids: natural ligands for an orphan nuclear receptor. Science 284:1365–1368
Parséus A, Sommer N, Sommer F et al (2017) Microbiota-induced obesity requires farnesoid X receptor. Gut 66:429–437
Pathak P, Liu H, Boehme S et al (2017) Farnesoid X receptor induces Takeda G-protein receptor 5 cross-talk to regulate bile acid synthesis and hepatic metabolism. J Biol Chem 292:11055–11069
Pathak P, Xie C, Nichols RG et al (2018) Intestine farnesoid X receptor agonist and the gut microbiota activate G-protein bile acid receptor-1 signaling to improve metabolism. Hepatology 68:1574–1588
Pencek R, Marmon T, Roth JD et al (2016) Effects of obeticholic acid on lipoprotein metabolism in healthy volunteers. Diabetes Obes Metab 18:936–940
Potthoff MJ, Boney-Montoya J, Choi M et al (2011) FGF15/19 regulates hepatic glucose metabolism by inhibiting the CREB-PGC-1α pathway. Cell Metab 13:729–738
Rao A, Kosters A, Mells JE et al (2016) Inhibition of ileal bile acid uptake protects against nonalcoholic fatty liver disease in high-fat diet–fed mice. Sci Transl Med 8:357ra122–357ra357
Raufman JP, Dawson PA, Rao A et al (2015) Slc10a2-null mice uncover colon cancer-promoting actions of endogenous fecal bile acids. Carcinogenesis 36:1193–1200
Renga B, Mencarelli A, Cipriani S et al (2013) The bile acid sensor FXR is required for immune-regulatory activities of TLR-9 in intestinal inflammation. PLoS One 8:e54472
Ryan KK, Tremaroli V, Clemmensen C et al (2014) FXR is a molecular target for the effects of vertical sleeve gastrectomy. Nature 509:183–188
Sayin SI, Wahlström A, Felin J et al (2013) Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab 17:225–235
Seok S, Fu T, Choi SE et al (2014) Transcriptional regulation of autophagy by an FXR-CREB axis. Nature 516:108–111
Shapiro H, Kolodziejczyk AA, Halstuch D et al (2018) Bile acids in glucose metabolism in health and disease. J Exp Med 215:383–396
Sjöberg BG, Straniero S, Angelin B et al (2017) Cholestyramine treatment of healthy humans rapidly induces transient hypertriglyceridemia when treatment is initiated. Am J Physiol Endocrinol Metab 313:E167–E174
Slijepcevic D, van de Graaf SFJ (2017) Bile acid uptake transporters as targets for therapy. Dig Dis 35:251–258
Spinelli V, Lalloyer F, Baud G et al (2016) Influence of Roux-en-Y gastric bypass on plasma bile acid profiles: a comparative study between rats, pigs and humans. Int J Obes 40:1260–1267
Stahl GE, Mascarenhas MR, Fayer JC et al (1993) Passive jejunal bile salt absorption alters the enterohepatic circulation in immature rats. Gastroenterology 104:163–173
Stenman LK, Holma R, Korpela R (2012) High-fat-induced intestinal permeability dysfunction associated with altered fecal bile acids. World J Gastroenterol 18:923–929
Stroeve JH, Brufau G, Stellaard F et al (2010) Intestinal FXR-mediated FGF15 production contributes to diurnal control of hepatic bile acid synthesis in mice. Lab Investig 90:1457–1467
Suchy FJ, Balistreri WF, Heubi JE et al (1981) Physiologic cholestasis: elevation of the primary serum bile acid concentrations in normal infants. Gastroenterology 80:1037–1041
Sun R, Yang N, Kong B et al (2017) Orally administered berberine modulates hepatic lipid metabolism by altering microbial bile acid metabolism and the intestinal FXR signaling pathway. Mol Pharmacol 91:110–122
Sun L, Xie C, Wang G et al (2018a) Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat Med 24:1919–1929
Sun L, Ma L, Ma Y et al (2018b) Insights into the role of gut microbiota in obesity: pathogenesis, mechanisms, and therapeutic perspectives. Protein Cell 9:397–403
Takahashi S, Fukami T, Masuo Y et al (2016) Cyp2c70 is responsible for the species difference in bile acid metabolism between mice and humans. J Lipid Res 57:2130–2137
Teixeira TF, Souza NC, Chiarello PG et al (2012) Intestinal permeability parameters in obese patients are correlated with metabolic syndrome risk factors. Clin Nutr 31:735–740
Temel RE, Brown JM (2015) A new model of reverse cholesterol transport: enTICEing strategies to stimulate intestinal cholesterol excretion. Trends Pharmacol Sci 36:440–451
Thomas C, Gioiello A, Noriega L et al (2009) TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab 10:167–177
Thompson CA, Wojta K, Pulakanti K et al (2017) GATA4 is sufficient to establish jejunal versus ileal identity in the small intestine. Cell Mol Gastroenterol Hepatol 24:422–446
Thulesen J (2004) Glucagon-like peptide 2 (GLP-2), an intestinotrophic mediator. Curr Protein Pept Sci 5:51–65
Toledo M, Batista-Gonzalez A, Merheb E et al (2018) Autophagy regulates the liver clock and glucose metabolism by degrading CRY1. Cell Metab 28:268–281
Tomlinson E, Fu L, John L et al (2002) Transgenic mice expressing human fibroblast growth factor-19 display increased metabolic rate and decreased adiposity. Endocrinology 143:1741–1747
Trabelsi M-S, Daoudi M, Prawitt J et al (2015) Farnesoid X receptor inhibits glucagon-like peptide-1 production by enteroendocrine L cells. Nat Commun 6:7629
Trauner M, Fuchs CD, Halilbasic E (2017) New therapeutic concepts in bile acid transport and signaling for management of cholestasis. Hepatology 65:1393–1404
Uranga RM, Keller JN (2010) Diet and age interactions with regards to cholesterol regulation and brain pathogenesis. Curr Gerontol Geriatr Res 2010:219683
van de Wiel SMW, de Waart DR, Oude Elferink RPJ et al (2018) Intestinal farnesoid X receptor activation by pharmacologic inhibition of the organic solute transporter α-β. Cell Mol Gastroenterol Hepatol 28:223–237
van Dijk TH, Grefhorst A, Oosterveer MH et al (2009) An increased flux through the glucose 6-phosphate pool in enterocytes delays glucose absorption in Fxr−/− mice. J Biol Chem 284:10315–10323
van Erpecum KJ, Schaap FG (2015) Intestinal failure to produce FGF19: a culprit in intestinal failure-associated liver disease? J Hepatol 62:1231–1233
Vaquero J, Monte MJ, Dominguez M et al (2013) Differential activation of the human farnesoid X receptor depends on the pattern of expressed isoforms and the bile acid pool composition. Biochem Pharmacol 86:926–939
Vavassori P, Mencarelli A, Renga B et al (2009) The bile acid receptor FXR is a modulator of intestinal innate immunity. J Immunol 183:6251–6261
Verbeke L, Farre R, Verbinnen B et al (2015) The FXR agonist obeticholic acid prevents gut barrier dysfunction and bacterial translocation in cholestatic rats. Am J Pathol 185:409–419
Verbeke L, Mannaerts I, Schierwagen R et al (2016) FXR agonist obeticholic acid reduces hepatic inflammation and fibrosis in a rat model of toxic cirrhosis. Sci Rep 16:33453
Verhulst PM, van der Velden LM, Oorschot V et al (2010) A flippase-independent function of ATP8B1, the protein affected in familial intrahepatic cholestasis type 1, is required for apical protein expression and microvillus formation in polarized epithelial cells. Hepatology 51:2049–2060
Voshol PJ, Schwarz M, Rigotti A et al (2001) Down-regulation of intestinal scavenger receptor class B, type I (SR-BI) expression in rodents under conditions of deficient bile delivery to the intestine. Biochem J 356:317–325
Wagner SA, Beli P, Weinert BT et al (2012) Proteomic analyses reveal divergent ubiquitylation site patterns in murine tissues. Mol Cell Proteomics 11:1578–1585
Walker EM, Thompson CA, Kohlnhofer BM et al (2014) Characterization of the developing small intestine in the absence of either GATA4 or GATA6. BMC Res Notes 11:902
Wang H, Chen J, Hollister K et al (1999) Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell 3:543–553
Wang Y-D, Chen W, Wang M et al (2008) Farnesoid X receptor antagonizes nuclear factor κB in hepatic inflammatory response. Hepatology 48:1632–1643
Wang XX, Luo Y, Wang D et al (2017) A dual agonist of farnesoid X receptor (FXR) and the G protein-coupled receptor TGR5, INT-767, reverses age-related kidney disease in mice. J Biol Chem 292:12018–12024
Watanabe M, Houten SM, Mataki C et al (2006) Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 439:484–489
Wheeler SG, Hammond CL, Jornayvaz FR et al (2014) Ostα−/− mice exhibit altered expression of intestinal lipid absorption genes, resistance to age-related weight gain, and modestly improved insulin sensitivity. Am J Physiol Gastrointest Liver Physiol 306:G425–G438
Williams JA, Thomas AM, Li G et al (2012) Tissue specific induction of p62/Sqstm1 by farnesoid X receptor. PLoS One 7:e43961
Wu Y, Aquino CJ, Cowan DJ et al (2013) Discovery of a highly potent, nonabsorbable apical sodium-dependent bile acid transporter inhibitor (GSK2330672) for treatment of type 2 diabetes. J Med Chem 56:5094–5114
Xie C, Jiang C, Shi J et al (2017) An intestinal farnesoid X receptor-ceramide signaling axis modulates hepatic gluconeogenesis in mice. Diabetes 66:613–626
Xiong X, Wang X, Lu Y et al (2014) Hepatic steatosis exacerbated by endoplasmic reticulum stress-mediated downregulation of FXR in aging mice. J Hepatol 60:847–854
Zhang Y, Kast-Woelbern HR, Edwards PA (2003) Natural structural variants of the nuclear receptor farnesoid X receptor affect transcriptional activation. J Biol Chem 278:104–110
Zhang Y, Castellani LW, Sinal CJ et al (2004) Peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α) regulates triglyceride metabolism by activation of the nuclear receptor FXR. Genes Dev 18:157–169
Zhang Y, Edwards PA (2008) FXR signaling in metabolic disease. FEBS Lett 582:10–18
Zhang L, Xie C, Nichols RG et al (2016) Farnesoid X receptor signaling shapes the gut microbiota and controls hepatic lipid metabolism. mSystems 1:e00070–e00016
Zhou M, Luo J, Chen M et al (2017) Mouse species-specific control of hepatocarcinogenesis and metabolism by FGF19/FGF15. J Hepatol 66:1182–1192
Zimmermann P, Hirsch-Hoffmann M, Hennig L et al (2004) GENEVESTIGATOR. Arabidopsis microarray database and analysis toolbox. Plant Physiol 136:2621–2632
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
van Zutphen, T. et al. (2019). Potential of Intestine-Selective FXR Modulation for Treatment of Metabolic Disease. In: Fiorucci, S., Distrutti, E. (eds) Bile Acids and Their Receptors. Handbook of Experimental Pharmacology, vol 256. Springer, Cham. https://doi.org/10.1007/164_2019_233
Download citation
DOI: https://doi.org/10.1007/164_2019_233
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-22004-4
Online ISBN: 978-3-030-22005-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)