
Abstract
Voltage-gated sodium channel (VGSC) beta (β) subunits have been called the “overachieving” auxiliary ion channel subunit. Indeed, these subunits regulate the trafficking of the sodium channel complex at the plasma membrane and simultaneously tune the voltage-dependent properties of the pore-forming alpha-subunit. It is now known that VGSC β-subunits are capable of similar modulation of multiple isoforms of related voltage-gated potassium channels, suggesting that their abilities extend into the broader voltage-gated channels. The gene family for these single transmembrane immunoglobulin beta-fold proteins extends well beyond the traditional VGSC β1–β4 subunit designation, with deep roots into the cell adhesion protein family and myelin-related proteins – where inherited mutations result in a myriad of electrical signaling disorders. Yet, very little is known about how VGSC β-subunits support protein trafficking pathways, the basis for their modulation of voltage-dependent gating, and, ultimately, their role in shaping neuronal excitability. An evolutionary approach can be useful in yielding new clues to such functions as it provides an unbiased assessment of protein residues, folds, and functions. An approach is described here which indicates the greater emergence of the modern β-subunits roughly 400 million years ago in the early neurons of Bilateria and bony fish, and the unexpected presence of distant homologues in bacteriophages. Recent structural breakthroughs containing α and β eukaryotic sodium channels containing subunits suggest a novel role for a highly conserved polar contact that occurs within the transmembrane segments. Overall, a mixture of approaches will ultimately advance our understanding of the mechanism for β-subunit interactions with voltage-sensor containing ion channels and membrane proteins.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Bacterial ion channels
- Evolution of membrane proteins and electrical signaling
- Ion channel auxiliary subunits
- NaV
- Sodium channel
- Voltage-gated ion channels
1 Sodium Channel Basics
Voltage-gated sodium channels (VGSC) are critical facilitators of electrical conduction in cardiovascular and neuronal tissues, as well as traditionally non-excitable cell types. The initial characterization of the VGSC protein complex identified two ~40 kDa proteins associated with the channel in a 1:1:1 ratio, termed the VGSC β auxiliary subunits. These β-subunits immunoprecipitated with the channel and were determined necessary for generating physiological current reproduction when the α-subunit is expressed heterologously (Hartshorne and Catterall 1981, 1984; Hartshorne et al. 1985). Since their initial identification, β-subunits have been shown to modulate VGSC gating and kinetics in addition to altering VGSC expression and pharmacology and further implications in disease phenotypes and nonconduction adhesion roles.
Four different β-subunits have been identified: β1, β2, β3, and β4 along with an alternative splice variant of β1 termed β1b (Isom et al. 1992, 1995a; Morgan et al. 2000; Yu et al. 2003; Qin et al. 2003). Membrane bound like the VGSC α-subunit, the single membrane-spanning segment of each β-subunit connects a large extracellular N-terminal domain to a short intracellular sequence. The exception to this topology is β1b whereby a skipped splice site prior to the β1 transmembrane exon results in a soluble protein. The large extracellular domain of the β-subunits is dominated by an immunoglobulin (Ig) fold of the V-set type that is highly glycosylated comprising nearly 30% of the proteins' molecular weight (Messner and Catterall 1985; Isom and Catterall 1996; Roberts and Barchi 1987). This Ig fold of β-subunits, which has similarity to the adhesion molecule contactin, targets binding for protein-protein interactions (Xiao et al. 1999). These β-subunits thus support a myriad of physiological roles, some of which are achieved through their tissue specific modulation of sodium channel expression and voltage-dependent gating. Other cellular effects appear to arise independently of their roles in tuning sodium channel electrical signaling behavior, these alternative roles rely heavily on the adhesion aspects of the β-subunit architecture.
Consistent with their primary effects on electrical signaling rhythms, β-subunits fine-tune the responses of the pore-forming α-subunit to transmembrane voltage. For instance, it was shown early on that co-expression of the neuronal channel Nav1.2 with β1 significantly accelerates the fast-inactivation process (Isom et al. 1992; Patton et al. 1994). Other VGSCs including Nav1.1–4, 1.6, and 1.7 display accelerated inactivation kinetics when β1 is co-expressed; adversely, Nav1.5 and Nav1.8 already have comparatively fast inactivation and have minimal if any inactivation modulation by β1 (Patton et al. 1994; Smith and Goldin 1998; Bennett et al. 1993; Wallner et al. 1993; Dietrich et al. 1998; Shcherbatko et al. 1999; Qu et al. 1995; Zhao et al. 2011). However, one complicating factor is that such effects on inactivation, and other channel gating modulatory effects, have varied between groups reporting results and between expression systems utilized, a possible result due to the presence of endogenous β-subunits in the expression systems (Isom et al. 1995b; Isom 2001; Chioni et al. 2009). Further, these so-called VGSC β-subunits have been recently shown to modulate multiple voltage-gated potassium channels (Deschênes and Tomaselli 2002; Marionneau et al. 2012; Nguyen et al. 2012a, b). Another striking effect the β-subunits have on VGSCs is to significantly increase current density; this effect is trafficking related resulting in channel upregulation at the plasma membrane (Kazarinova-Noyes et al. 2001). This increased presence of the VGSC at the plasma membrane allows for quicker conduction which combined with an hastened inactivation rate produces a faster return to baseline after action potential firing allowing for more rapid subsequent electrical stimulation.
β-subunits are principally located in excitable tissues (muscle and neurons) where their interaction can tune the VGSC conduction in a tissue-specific manner. While the β-subunits primary role appears to be electrically related, the do not have a direct role in the movement of ions across the plasma membrane and can be found in non-excitable tissues such as glial astrocytes, Schwann cells, and in the kidney where the β-subunit function is theorized to be adhesion focused (Oh and Waxman 1994; Isom 2002). The β-subunits are differently regulated by tissues and within a tissue, for instance, the heart has β1, β2, and β3 at the transverse-tubules and β1, β2, and β4 at the intercalated disks; β1 is found at both locations with the Y181 phosphorylation modified β1 trafficking to the ICD, while unphosphorylated proceeds at the T-tubules (Maier et al. 2004; Malhotra et al. 2004). Further evidence of subcellular regulation is the increased density of β-subunits at the nodes of Ranvier where the β-subunits can interact with VGSC to alter gating, this nodal regulation has been linked to β2 interaction with tenascin-R and tenascin-C; however, β-subunit association with nodes also requires both ankyrin and VSGCs to localize (Xiao et al. 1999; Chen et al. 2012; Srinivasan et al. 1998). β-subunit interaction with VGSC is not limited to electrophysiological modulation; expression of β1 or β3 in HEK cells alters trafficking and glycosylation content depending on the β-subunit associated (Laedermann et al. 2013). Co-immunoprecipitation analysis have identified ankyrin-G, contactin, NrCAM, NF155, and NF186 as interaction partners with β-subunits with differing specificity for each subunit and the subunit post-translational modifications (Kazarinova-Noyes et al. 2001; Malhotra et al. 2000, 2004; Ratcliffe et al. 2001; McEwen and Isom 2004). β-subunits not only have a similar Ig domain to myelin protein zero, the major constituent of the PNS myelin sheath the β-subunits also have roles in neuronal development: β-subunit axonal guidance is experimentally observed by an increased neurite length when grown on a β1 supporting monolayer; β2 nor β4 have similar effects in the supporting cells; however, increased neuronal β4 expression promotes neurite elongation (Shapiro et al. 1996; Davis et al. 2004; Zhou et al. 2012). β1 neurite pathfinding effects require the adhesion molecule contactin and Fyn kinase, suggesting lipid raft localization; the extracellular domain of β1 is sufficient to mediate this effect as β1B has similar properties (Brackenbury et al. 2008; Patino et al. 2011). Lipid raft localization is where the β-subunits are targeted by proteases including the gamma secretase complex and BACE1, that release the extracellular Ig domain and intracellular domain of the β-subunits, thus expanding the β-subunit interactions from the membrane to surrounding cells and intracellular locations (Patino et al. 2011; Kim et al. 2005, 2007; Wong et al. 2005).
2 VGSC and Human Disease
Epileptic seizures are linked to elevated BACE1 activity in early- and late-onset Alzheimer’s disease, this electrical disruption is partially contributed from increased Nav1.1 surface expression, a result of the BACE1 mediated β2 intracellular domain release (Kim et al. 2007). β-subunits have been implicated more directly with other electrical disorders in humans, principally the epileptic disorders (SIDS, GEFS+, or Dravet syndrome) and cardiac disorders (Brugada syndrome, long QT syndrome, atrial and ventricular fibrillation) with known clinical mutations of β1 (R85C/H, E87Q, I106F, C121W, R125C/L, D153N, W179X, and a splice site mutation) (Scheffer et al. 2007; Xu et al. 2007; Watanabe et al. 2008, 2009; Ogiwara et al. 2012; Wallace et al. 1998; Patino et al. 2009; Fendri-Kriaa et al. 2011; Audenaert et al. 2003), β1B (H162P, P213T, R214Q, and G257R) (Patino et al. 2011; Hu et al. 2012; Yuan et al. 2014; Riuró et al. 2014), β2 (R28Q/W and D211G) (Watanabe et al. 2009; Riuró et al. 2013), β3 (R6K, L10P, V36M, V54G, V110I, A130V, and M161T) (Tan et al. 2010; Hu et al. 2009; Valdivia et al. 2009; Ishikawa et al. 2013; Wang et al. 2010), and β4 (V162G, I166L, L179F, and S206L) (Tan et al. 2010; Li et al. 2013; Medeiros-Domingo et al. 2007). These mutations are found throughout the β-subunits domains, with effects ranging from nonfunctional proteins to altered interaction with VGSCs. Null mice for the four β-subunits have been produced with differing effects. β1 has a critical role in neuronal and cardiac function, this is exhibited by β1 null effects including developmental abnormalities, perturbed axonal pathfinding, spontaneous seizures, and extended QT intervals (Brackenbury et al. 2008, 2013; Chen et al. 2004; Lopez-Santiago et al. 2007). However, other β-subunit null mice have less drastic effects. For instance, β2 are prone to seizures, β3 have arrhythmic tendencies, and β4 have balance and motor defects, none of which are as serious as the β1 effects, suggesting a less critical role or adequate compensation by other proteins (Chen et al. 2002; Hakim et al. 2008; Ransdell et al. 2017). β-subunits alter the channel pharmacology, natural drugs including the toxins STX, and μ-conotoxins have altered binding when expressed with different β-subunits. In particular, the cone snail venom μO-conotoxin MrVIB has significantly improved block on Nav1.8 with any of the four β-subunit co-expressed compared to the channel alone and differing degrees of block depending on the interacting β-subunit (Schmidt et al. 1985; Wilson et al. 2011, 2015; Zhang et al. 2013). These pharmacological effects are extended to clinical drugs which act through VGSCs. The antiarrhythmic Lidocaine has over a two-fold decrease in affinity for Nav1.5 when expressed with β1, and the antiepileptic drug carbamazepine is incapable of blocking repetitive action potential firing without β1 (Makielski et al. 1996; Uebachs et al. 2012). The adhesion properties of β-subunits make them a strong candidate for effects in cancer metastases, β-subunits have been impacted in tumor migration and invasiveness and further association to angiogenesis has been observed in tumors originating from breast, cervical, glioblastoma, NSCL, and prostate cancers (Chioni et al. 2009; Diss et al. 2008; Aronica et al. 2003; Nelson et al. 2014; Brackenbury 2012).
3 β-Subunit Homology from the Perspective of Primary Sequence
At the sequence level, the β-subunits are diverse. The human VGSC α-subunits range from 53 to 88% identity; within the β-subunits, β1 and β3 are the most similar with nearly 50% identity, subsequently the second most similar are β2 and β4 with only 27% identity, and the other comparisons come in at less than 25% identity. β2 and β4 were determined via alanine scanning to bind α-channels via a disulfide bridge at Cys55 and Cys58, respectively, that separate under reducing conditions (Chen et al. 2012; Buffington and Rasband 2013). Similar scanning of Nav1.2 determined Cys910 of Nav1.2 DIIS5–S6 to be the β-subunit intermolecular disulfide bonding pair (Das et al. 2016). However, not all VGSC have this conserved cysteine for covalent attachment, such as Nav1.5 the cardiac isoform which does not covalently bind with either β2 or β4.
Unlike β2 and β4, the β1-subunit is not covalently bound; if theorizing a 1:1:1 ratio of the α:β1:β2 and that β1 has a direct interaction with α, then β1 must be interacting at a differing location than β2. The extracellular domains of the β-subunits are important in their interaction, nodal localization is interrupted by the Ig domain mutant of β2 C55A that breaks the disulfide bond; the extracellular Ig loop of β1 has similar importance on VGSC interaction, evidence by a GPI-linked β1 extracellular domain can recapitulate the β1 modulatory effects (Chen et al. 2012; Makita et al. 1996a; McCormick et al. 1998). Studies making chimeras using either Nav1.2 or Nav1.4 which have significant modulation by β1 and Nav1.5 that is not modulated by β1 have identified extracellular locations of the α required for modulation (Makita et al. 1996a, b; Qu et al. 1999). Further evidence for extracellular interaction has been recently identified with voltage-clamp fluorometry, this data depicts β1 and β3 interacting with the voltage-sensing domains of Nav1.5 and suggests a close proximity of the non-covalently bound β-subunit to the channel (Sharkey et al. 1984; Zhu et al. 2017).
4 Evolutionary History of Beta-Subunits
All VGSC β-subunits are type I integral membrane proteins, i.e., they show a single transmembrane with a cytosolically located C-terminus. The extracellular V-set Ig domain is a Greek-key beta-sandwich structure resembling the antibody variable domain, which is connected to the transmembrane alpha helical segment through a neck domain (Namadurai et al. 2015). V-set domains are members of a large class of domains, namely, immunoglobulin-like domains, ubiquitously present in all kingdoms of life (Bork et al. 1994). This fold is characterized by a two-layer sandwich of seven to nine antiparallel β-strands arranged in two planes of β-sheets.
The origin of immunoglobulin-like folds has long been debated. The traditionally favored hypothesis posits that this fold is a thermodynamically favored “platonic form” toward which proteins of a given length would spontaneously converge (Lesk and Chothia 1982). The observed distribution of immunoglobulin-like domain would then be the result of convergent evolution. A second hypothesis (that is gaining acceptance), instead, places emphasis on the biological function shared by a large portion of this superfamily: many members, though not all, are involved in the processes of cell adhesion, an observation suggesting a possible origin for the evolutionary conservation of the structure. However, despite the reasonability of this scenario, sequence conservation is poor across kingdoms and therefore evolutionary relatedness is difficult to establish with an acceptable degree of statistical confidence (Namadurai et al. 2015).
In contrast to other members of this superfamily, V-set domains are found almost exclusively in metazoan Bilateria, with a few occurrences in cnidarians. A notable exception to this monophyletic distribution in present in viruses: some poxyviruses contain V-set domains in their hemagglutinin and glycoprotein genes, a possible result of horizontal gene transfer events (Dermody et al. 2009). Whether these horizontal gene transfer events took place after the emergence of animals or before is still an open question. Indeed, the second largest group of viruses showing immunoglobulin-like domains are dsDNA bacteriophages. This observation raises the hypothesis that bacteriophages have played a crucial role in enabling extensive horizontal gene transfer events in bacteria and, possibly, in early eukaryotes (Fraser et al. 2007).
The evolutionary history of V-set domains constrains the possible evolutionary emergence events of the auxiliary β-subunits. In effect, demonstrating that VGSC β-subunits could not have appeared in their current form prior to the emergence of animals. This raises immediately a question: were β-subunits a response to the merging nervous systems in animals? Intriguingly, some occurrences of the V-set domain are found in the phylum of sponges, indicating that structural templates for β-subunits might have been available before nerve cells appeared. However, homologues of β-subunits found by scouring the entire UniProt database suggest that membrane-bound V-set domains might have been co-opted into the Nav auxiliary subunits role more recently than the emergence of excitable cells, suggesting their emergence was timed as animal nervous systems cultivated complexity. Notably, all genes containing detectable homology with human β-subunits are in vertebrata (Fig. 1). In particular, when pinpointed on the tree of life, these genes are found in bony fish ogranisms (Osteichthyes), which are in turn divided into the ray-finned fish (Actinopterygii) and lobe-finned fish (Sarcopterygii), but not in cartilaginous fish ogramisms (Chondrichthyes) like sharks. Fossil records help locate the splitting between Osteichthyes and Chondrichthyes at about 420 million years ago; thus, the present form of VGSC β-subunits, appeared in organisms already possessing a fully developed nervous system.

Phylogenetic structure of the genes homologous to SCN1B. The dendrogram shows the major branching and their statistical significance (support). Numbers greater than 0.7 indicate a large degree of confidence in the tree structure. Groups are labeled according to the annotated genes contained in each group. Note that several myelin-associated proteins are identified as SCN1B homologues
The evolution of the β-subunits can be best appreciated by analyzing the functionally homogenous families of β1 and β3. Since both families are present in ray-finned fish (Actinopterygii) and in lobe-finned fish (Sarcopterygii, ancestors of, among others, Reptilia and Mammalia), we can conclude that the common ancestor was present at the emergence of Euteleostomi in Silurian age, ca 420 Mya (Fig. 2). Consistently, the gene from Latimeria, which is considered the oldest representative of Sarcopterygii, is found close to the node separating the two main branches. Notably, the β1 and β3 branches show very similar organizations, completely consistent with the phylogenetic tree of Vertebrata. Since no homologous sequence from sharks has been detected, it is unlikely that the origin of these families can be further pinpointed.

Phylogenetic relationships between SCN1B and SCN3B genes. The dendrogram is restricted to the branches containing the annotated SCN1B and SCN3B genes. Note that the structure is the same for the two major branches and is consistent with known cladograms
An interesting insight that can be gained from this database-wide search for β-subunits homologues, is an appreciation that the overall size of the β-subunit superfamily extends well beyond the traditional β1–β4 gene nomenclature. The dendrogram shown in Fig. 1 highlights the major branches, or clusters, in which these homologous genes are organized. Labels have been added to all the groups containing sequences with experimentally validated annotation (i.e., belonging to the Swiss-Prot database). A first notable feature that emerges from this representation is that β-subunits are formed by two major groups: the first one consists of the β1 and β3 family, while the second contains the β2 and β4 families. Importantly, this phylogenetic organization of the four families is consistent with the observed functional differences (Namadurai et al. 2015). The second interesting feature conveyed by the three is the presence of several intervening groups of genes labeled as JAML, MYP0, MPZL1, MPZL2, and MPZL1. The position in the tree suggests that these groups are more similar to the members of the β2 and β4 groups than those of the β1 and β3 one. Is it then possible that some of the genes present in these phylogenetic branches encode for a thus far unknown auxiliary subunit? Intriguing insights into a possible involvement of ion channel regulation by these genes come from the identified clinical variants. For instance, the gene group labeled MYP0 contains the human gene encoding for myelin protein zero, a glycoprotein that is a crucial structural component of the myelin sheath. Mutations in this protein are associated with the diseases Charcot-Marie-Tooth and Dejerine-Sottas. In most of their forms, these diseases are associated with demyelination. However, some forms such as Charcot-Marie-Tooth type II disease is associated with mutations in MYP0 that are not demyelinating neuropathies, despite the fact that they result in altered motor action potentials (Chapon et al. 1999). Thus, it is tempting to speculate that, besides their known cellular function, genes like MYP0 can have a more functional interaction with neuronal VGSCs.
5 Structural Features and Regions of Sequence Conservation
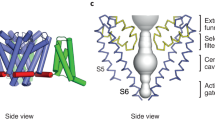
A recently determined structure of Nav1.4 from the electric eel, obtained through cryo-electron microscopy, provides some unique insights into possible mechanisms of action of the regulatory subunits (Yan et al. 2017). Presence of an interacting partner for Nav1.4 in the electron density was identified as β1. An interesting feature revealed by this structural model is the fact that the α- and β-subunits interact through both their transmembrane and extracellular domains (Fig. 3a). This was somewhat unanticipated given that previous structure-function studies suggested a predominate role of the β-subunit extracellular Ig domain such the transmembrane segment was predicted as a more generic interaction interface. Interestingly, β1 establishes extensive interactions with structural elements from three distinct domains from the sodium channel α-subunit: DI, DIII, and DIV. In particular, the extracellular part of β1 shows contacts with loop 5 from DI and loop 6 from DIV, in addition to a salt bridge with R1028 located on the S1–S2 loop of DIII. Besides these interactions between the two solvent exposed regions, β1 and Nav1.4 show a remarkably large number of residue-residue contacts in their hydrophobic transmembrane sections (Fig. 3b). The protein-protein interaction interface involves voltage sensor segments S0 and S2 of DIII and is mostly composed by interweaving bulky hydrophobic side chains.

Structure of the eel β1/Nav1.4 complex as determined by cryo-electron microscopy. (a) Cartoon representation of the complex showing β1 (red), the voltage-sensor domains (blue), the pore domain (white), the L5 loop (cyan), and the L6 loop (yellow). The green dashed line highlights the transmembrane section of the protein-protein complex constituted by the voltage-sensor domain of DIII and the transmembrane part of β1. (b, c) Close-up of the transmembrane region of the β1/Nav1.4 interaction surface. The gray shading shows the molecular surface of the two interacting partners; a space filling representation is used to highlight the nonpolar (b) and polar (c) side chains at the protein-protein interface
A particularly intriguing set of residue-residue interactions between β1 and Nav1.4 are those involving polar side chains (Fig. 3c). As noted previously (Namadurai et al. 2015), the polar residues of β1 and β3 within the transmembrane segments act as potential oligomerization regions between β-subunits. Intriguingly, these polar interaction side chains are involved in seemingly specific recognition between the α- and the β-subunits. Particularly relevant is the hydrogen bond between Q174 from β1 and Y1043 from S2 of DIII: in spite of their marked polar character, these two side chains are located in the middle of the membrane, i.e., in a region completely inaccessible to water molecules and typically devoid of hydrogen bond donors or acceptors. The net free energy gain for establishing such interaction in this environment is typically large (between 2 and 5 kcal/mol); therefore, glutamine (and to a lesser extent asparagines) residue is often responsible for multimerization of transmembrane helices (Choma et al. 2000). In the context of the α/β complex, this residue-residue interaction may provide a structural determinant of the affinity between β1 and Nav1.4 and possibly determines the selectivity against other class of β-subunits like β2 and β4. In addition to the Q174-Y1043 contact, there are two other polar interactions involving the two proteins occurring between side chains located in the interfacial region of the lipid bilayer. The first one is established between Y167 from β1 and K1039 from S2, possibly established with an intervening water molecule or lipid head group. The second polar interaction involves an entire cluster of polar side chains from β1 (R155, S159, and E163) and the phenyl group of Y1036 on S2, whose hydroxyl group seemingly donates an H-bond to the carboxylate of E163 and accepts two H-bonds, one from R155 and the other from S159.
In light of the specific contacts highlighted by the cryo-EM structure, it is interesting to analyze the sequence conservation pattern of distinct β-subunits within multiple species. In particular, from a database-wide search for β-subunits homologues across all the sequenced organisms, one can characterize the degree of sequence conservation at each structural position and from this infer which of the contacts observed in the β1/Nav1.4 complex are crucial and have been thus preserved across evolution. The first interesting insights emerging from this analysis is that the transmembrane domain is one of the most conserved regions of the β-subunits for both the β1 and β3 and the β2 and β4 groups (Fig. 4a, b). The second interesting observation is that the polar amino acids involved in the interaction with S2 of Nav1.4 DIII appear to be strongly conserved (Fig. 4a). Importantly, amino acids corresponding to Q174 in the electric eel β1 are invariably polar: at this position, only glutamine or threonine side chains are observed in other species. An even stronger sequence conservation is observed at the positions corresponding to S159, E163, and Y167 of the electric eel β1. Importantly, the remaining conserved amino acids are also found to establish hydrophobic interactions in the β1/Nav1.4; these are M166, I170, and L177. As a result, a polar/nonpolar alternate pattern of conservation is apparent in the β1 and β3 family. Intriguingly, this pattern is absent in the β2 or β4 with the exception of a conserved serine amino acid, the side chains are mostly nonpolar. While it is impossible to draw strong conclusions from this information, it is tempting to speculate that the different functional role of β2 and β4 with respect to β1 and β3 might result from these differences in sequence, which, ultimately might result in a different binding mode to the α-subunit.

Sequence conservation profile for the SCN1B/SCN3B (a) and SCN2B/SCNB4 (b) families. Shown are sequence logos: a concise graphical representation of sequence multiple sequence alignments. Amino acid one letter code is used to show the residues present in each position of the sequence. The height of each font is proportional to the frequency of occurrence of each amino acid, while the height of the entire column indicates the overall sequence conservation at a given position. The yellow shaded boxes highlight the transmembrane regions
In conclusion, the overall understanding of voltage-gated sodium channel auxiliary subunits has benefited from a variety of biochemical, electrophysiological, and structural characterizations. An evolutionary analysis suggests homologues exist in viral bacteriophage proteins, with the modern manifestation of the beta-subunit gene family emerging within the existing nervous system of bony fish ~420 myo. Thus, it is clear that the VGSC β-subunit family has deeper roots in the foundation of neurobiology and, possibly, playing a supporting role in the formation of the nervous system in animals. Given their widespread expression, and multiple modulatory targets within excitable cells, they are associated with a plethora of human diseases. The advent of recent structural breakthroughs are beginning to provide a framework for how this class of diverse auxiliary subunit might regulate the voltage-dependent properties of ion channels and may herald a new era of efforts to guide structure-based therapeutics.
References
Aronica E et al (2003) Expression and regulation of voltage-gated sodium channel β1 subunit protein in human gliosis-associated pathologies. Acta Neuropathol 105(5):515–523
Audenaert D et al (2003) A deletion in SCN1B is associated with febrile seizures and early-onset absence epilepsy. Neurology 61:854–856
Bennett PB Jr, Makita N, George AL Jr (1993) A molecular basis for gating mode transitions in human skeletal muscle Na+ channels. FEBS Lett 326(1–3):21–24
Bork P, Holm L, Sander C (1994) The immunoglobulin fold. Structural classification, sequence patterns and common core. J Mol Biol 242(4):309–320
Brackenbury WJ (2012) Voltage-gated sodium channels and metastatic disease. Channels 6(5):352–361
Brackenbury WJ et al (2008) Voltage-gated Na+ channel β1 subunit-mediated neurite outgrowth requires Fyn kinase and contributes to postnatal CNS development in vivo. J Neurosci 28(12):3246–3256
Brackenbury WJ et al (2013) Abnormal neuronal patterning occurs during early postnatal brain development of Scn1b-null mice and precedes hyperexcitability. Proc Natl Acad Sci 110(3):1089–1094
Buffington SA, Rasband MN (2013) Na+ channel-dependent recruitment of Navβ4 to axon initial segments and nodes of Ranvier. J Neurosci 33(14):6191–6202
Chapon F et al (1999) Axonal phenotype of Charcot-Marie-Tooth disease associated with a mutation in the myelin protein zero gene. J Neurol Neurosurg Psychiatry 66(6):779–782
Chen C et al (2002) Reduced sodium channel density, altered voltage dependence of inactivation, and increased susceptibility to seizures in mice lacking sodium channel β2-subunits. Proc Natl Acad Sci 99(26):17072–17077
Chen C et al (2004) Mice lacking sodium channel β1 subunits display defects in neuronal excitability, sodium channel expression, and nodal architecture. J Neurosci 24(16):4030–4042
Chen C et al (2012) Identification of the cysteine residue responsible for disulfide linkage of Na+ channel α and β2 subunits. J Biol Chem 287(46):39061–39069
Chioni A-M et al (2009) A novel adhesion molecule in human breast cancer cells: voltage-gated Na+ channel β1 subunit. Int J Biochem Cell Biol 41(5):1216–1227
Choma C et al (2000) Asparagine-mediated self-association of a model transmembrane helix. Nat Struct Biol 7(2):161–166
Das S et al (2016) Binary architecture of the Nav1.2-β2 signaling complex. eLife 5:e10960
Davis TH, Chen C, Isom LL (2004) Sodium channel β1 subunits promote neurite outgrowth in cerebellar granule neurons. J Biol Chem 279(49):51424–51432
Dermody TS et al (2009) Immunoglobulin superfamily virus receptors and the evolution of adaptive immunity. PLoS Pathog 5(11):e1000481
Deschênes I, Tomaselli GF (2002) Modulation of Kv4.3 current by accessory subunits. FEBS Lett 528(1–3):183–188
Dietrich PS et al (1998) Functional analysis of a voltage-gated sodium channel and its splice variant from rat dorsal root ganglia. J Neurochem 70:2262–2272
Diss J et al (2008) [Beta]-subunits of voltage-gated sodium channels in human prostate cancer: quantitative in vitro and in vivo analyses of mRNA expression. Prostate Cancer Prostatic Dis 11(4):325
Fendri-Kriaa N et al (2011) New mutation c.374C>T and a putative disease-associated haplotype within SCN1B gene in Tunisian families with febrile seizures. Eur J Neurol 18(5):695–702
Fraser JS, Maxwell KL, Davidson AR (2007) Immunoglobulin-like domains on bacteriophage: weapons of modest damage? Curr Opin Microbiol 10(4):382–387
Hakim P et al (2008) Scn3b knockout mice exhibit abnormal ventricular electrophysiological properties. Prog Biophys Mol Biol 98(2):251–266
Hartshorne RP, Catterall WA (1981) Purification of the saxitoxin receptor of the sodium channel from rat brain. Proc Natl Acad Sci 78(7):4620–4624
Hartshorne RP, Catterall WA (1984) The sodium channel from rat brain. Purification and subunit composition. J Biol Chem 259(3):1667–1675
Hartshorne RP et al (1985) Functional reconstitution of the purified brain sodium channel in planar lipid bilayers. Proc Natl Acad Sci U S A 82:240–244
Hu D et al (2009) A mutation in the β3 subunit of the cardiac sodium channel associated with Brugada ECG phenotype. Circ Cardiovasc Genet 2:270–278
Hu D et al (2012) A novel rare variant in SCN1Bb linked to Brugada syndrome and SIDS by combined modulation of Nav1.5 and Kv4.3 channel currents. Heart Rhythm 9(5):760–769
Ishikawa T et al (2013) Novel SCN3B mutation associated with Brugada syndrome affects intracellular trafficking and function of Nav1. 5. Circ J 77(4):959–967
Isom LL (2001) Sodium channel β subunits: anything but auxiliary. Neuroscientist 7(1):42–54
Isom LL (2002) The role of sodium channels in cell adhesion. Front Biosci 7(1):12–23
Isom LL, Catterall WA (1996) Na+ channel subunits and Ig domains. Nature 383(6598):307–308
Isom LL et al (1992) Primary structure and functional expression of the β subunit of the rat brain sodium channel. Science 256(5058):839–842
Isom LL et al (1995a) Structure and function of the β2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM motif. Cell 83(3):433–442
Isom LL et al (1995b) Functional co-expression of the 1 and type IIA subunits of sodium channels in a mammalian cell line. J Biol Chem 270(7):3306–3312
Kazarinova-Noyes K et al (2001) Contactin associates with Na+ channels and increases their functional expression. J Neurosci 21(19):7517–7525
Kim DY et al (2005) Presenilin/gamma-secretase-mediated cleavage of the voltage-gated sodium channel beta2-subunit regulates cell adhesion and migration. J Biol Chem 280:23251–23261
Kim DY et al (2007) BACE1 regulates voltage-gated sodium channels and neuronal activity. Nat Cell Biol 9(7):755–764
Laedermann CJ et al (2013) β1-and β3-voltage-gated sodium channel subunits modulate cell surface expression and glycosylation of Nav1. 7 in HEK293 cells. Front Cell Neurosci 7:137
Lesk AM, Chothia C (1982) Evolution of proteins formed by beta-sheets. II. The core of the immunoglobulin domains. J Mol Biol 160(2):325–342
Li RG et al (2013) Mutations of the SCN4B-encoded sodium channel β4 subunit in familial atrial fibrillation. Int J Mol Med 32(1):144–150
Lopez-Santiago LF et al (2007) Sodium channel SCN1B null mice exhibit prolonged QT and RR intervals. J Mol Cell Cardiol 43(5):636–647
Maier SKG et al (2004) Distinct subcellular localization of different sodium channel α and β subunits in single ventricular myocytes from mouse heart. Circulation 109(11):1421–1427
Makielski JC et al (1996) Coexpression of beta 1 with cardiac sodium channel alpha subunits in oocytes decreases lidocaine block. Mol Pharmacol 49(1):30–39
Makita N, Bennett PB, George AL (1996a) Multiple domains contribute to the distinct inactivation properties of human heart and skeletal muscle Na+ channels. Circ Res 78(2):244–252
Makita N, Bennett PB Jr, George AL Jr (1996b) Molecular determinants of B1 subunit-induced gating modulation in voltage-dependent Na+ channels. J Neurosci 16(22):7117–7127
Malhotra JD et al (2000) Sodium channel β subunits mediate homophilic cell adhesion and recruit ankyrin to points of cell-cell contact. J Biol Chem 275(15):11383–11388
Malhotra JD et al (2004) Tyrosine-phosphorylated and nonphosphorylated sodium channel β1 subunits are differentially localized in cardiac myocytes. J Biol Chem 279(39):40748–40754
Marionneau C et al (2012) The sodium channel accessory subunit Navbeta1 regulates neuronal excitability through modulation of repolarizing voltage-gated K(+) channels. J Neurosci 32(17):5716–5727
McCormick KA et al (1998) Molecular determinants of Na+ channel function in the extracellular domain of the β1 subunit. J Biol Chem 273(7):3954–3962
McEwen DP, Isom LL (2004) Heterophilic interactions of sodium channel β1 subunits with axonal and glial cell adhesion molecules. J Biol Chem 279(50):52744–52752
Medeiros-Domingo A et al (2007) SCN4B-encoded sodium channel β4 subunit in congenital long-QT syndrome. Circulation 116(2):134–142
Messner DJ, Catterall WA (1985) The sodium channel from rat brain. Separation and characterization of subunits. J Biol Chem 260(19):10597–10604
Morgan K et al (2000) β3: an additional auxiliary subunit of the voltage-sensitive sodium channel that modulates channel gating with distinct kinetics. Proc Natl Acad Sci 97(5):2308–2313
Namadurai S et al (2015) A new look at sodium channel β subunits. Open Biol 5(1):140192
Nelson M et al (2014) The sodium channel β1 subunit mediates outgrowth of neurite-like processes on breast cancer cells and promotes tumour growth and metastasis. Int J Cancer 135(10):2338–2351
Nguyen HM et al (2012a) Modulation of voltage-gated K+ channels by the sodium channel β1 subunit. Proc Natl Acad Sci 109(45):18577–18582
Nguyen HM et al (2012b) Modulation of Kv1 voltage-gated potassium channels by sodium channel beta subunits. Biophys J 102(3, Suppl 1):687a
Ogiwara I et al (2012) A homozygous mutation of voltage-gated sodium channel β1 gene SCN1B in a patient with Dravet syndrome. Epilepsia 53(12):e200–e203
Oh Y, Waxman SG (1994) The beta 1 subunit mRNA of the rat brain Na+ channel is expressed in glial cells. Proc Natl Acad Sci U S A 91:9985–9989
Patino GA et al (2009) A functional null mutation of SCN1B in a patient with Dravet syndrome. J Neurosci 29(34):10764–10778
Patino GA et al (2011) Voltage-gated Na(+) channel β1B: a secreted cell adhesion molecule involved in human epilepsy. J Neurosci 31(41):14577–14591
Patton DE et al (1994) The adult rat brain β1 subunit modifies activation and inactivation gating of multiple sodium channel alpha subunits. J Biol Chem 269(26):17649–17655
Qin N et al (2003) Molecular cloning and functional expression of the human sodium channel β1B subunit, a novel splicing variant of the β1 subunit. Eur J Biochem 270(23):4762–4770
Qu Y et al (1995) Modulation of cardiac Na+ channel expression in Xenopus oocytes by β1 subunits. J Biol Chem 270(43):25696–25701
Qu Y et al (1999) Functional roles of the extracellular segments of the sodium channel α subunit in voltage-dependent gating and modulation by β1 subunits. J Biol Chem 274(46):32647–32654
Ransdell JL et al (2017) Loss of Navβ4-mediated regulation of sodium currents in adult Purkinje neurons disrupts firing and impairs motor coordination and balance. Cell Rep 19(3):532–544
Ratcliffe CF et al (2001) Sodium channel β1 and β3 subunits associate with neurofascin through their extracellular immunoglobulin-like domain. J Cell Biol 154(2):427–434
Riuró H et al (2013) A missense mutation in the sodium channel β2 subunit reveals SCN2B as a new candidate gene for Brugada syndrome. Hum Mutat 34(7):961–966
Riuró H et al (2014) A missense mutation in the sodium channel β1b subunit reveals SCN1B as a susceptibility gene underlying long QT syndrome. Heart Rhythm 11(7):1202–1209
Roberts RH, Barchi RL (1987) The voltage-sensitive sodium channel from rabbit skeletal muscle. Chemical characterization of subunits. J Biol Chem 262(5):2298–2303
Scheffer IE et al (2007) Temporal lobe epilepsy and GEFS+ phenotypes associated with SCN1B mutations. Brain 130(1):100–109
Schmidt J, Rossie S, Catterall WA (1985) A large intracellular pool of inactive Na channel alpha subunits in developing rat brain. Proc Natl Acad Sci 82(14):4847–4851
Shapiro L et al (1996) Crystal structure of the extracellular domain from P0, the major structural protein of peripheral nerve myelin. Neuron 17:435–449
Sharkey RG, Beneski DA, Catterall WA (1984) Differential labeling of the α and β1 subunits of the sodium channel by photoreactive derivatives of scorpion toxin. Biochemistry 23(25):6078–6086
Shcherbatko A et al (1999) Voltage-dependent sodium channel function is regulated through membrane mechanics. Biophys J 77(4):1945–1959
Smith RD, Goldin AL (1998) Functional analysis of the rat I sodium channel in Xenopus oocytes. J Neurosci 18(3):811–820
Srinivasan J, Schachner M, Catterall WA (1998) Interaction of voltage-gated sodium channels with the extracellular matrix molecules tenascin-C and tenascin-R. Proc Natl Acad Sci U S A 95(26):15753–15757
Tan B-H et al (2010) Sudden infant death syndrome–associated mutations in the sodium channel beta subunits. Heart Rhythm 7(6):771–778
Uebachs M et al (2012) Loss of β1 accessory Na+ channel subunits causes failure of carbamazepine, but not of lacosamide, in blocking high-frequency firing via differential effects on persistent Na+ currents. Epilepsia 53(11):1959–1967
Valdivia CR et al (2009) Loss-of-function mutation of the SCN3B-encoded sodium channel β3 subunit associated with a case of idiopathic ventricular fibrillation. Cardiovasc Res 86(3):392–400
Wallace RH et al (1998) Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel sz1 subunit gene SCN1B. Nat Genet 19(4):366–370
Wallner M et al (1993) Modulation of the skeletal muscle sodium channel α-subunit by the β1-subunit. FEBS Lett 336(3):535–539
Wang P et al (2010) Functional dominant-negative mutation of sodium channel subunit gene SCN3B associated with atrial fibrillation in a Chinese GeneID population. Biochem Biophys Res Commun 398(1):98–104
Watanabe H et al (2008) Sodium channel β1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J Clin Invest 118(6):2260–2268
Watanabe H et al (2009) Mutations in sodium channel β1- and β2-subunits associated with atrial fibrillation. Circ Arrhythm Electrophysiol 2(3):268–275
Wilson MJ et al (2011) Navβ subunits modulate the inhibition of Nav1.8 by the analgesic gating modifier μO-conotoxin MrVIB. J Pharmacol Exp Ther 338(2):687–693
Wilson MJ et al (2015) Α-and β-subunit composition of voltage-gated sodium channels investigated with μ-conotoxins and the recently discovered μO §-conotoxin GVIIJ. J Neurophysiol 113(7):2289–2301
Wong H-K et al (2005) β subunits of voltage-gated sodium channels are novel substrates of β-site amyloid precursor protein-cleaving enzyme (BACE1) and γ-secretase. J Biol Chem 280(24):23009–23017
Xiao Z-C et al (1999) Tenascin-R is a functional modulator of sodium channel β subunits. J Biol Chem 274(37):26511–26517
Xu R et al (2007) Generalized epilepsy with febrile seizures plus–associated sodium channel β1 subunit mutations severely reduce β subunit–mediated modulation of sodium channel function. Neuroscience 148(1):164–174
Yan Z et al (2017) Structure of the Nav1.4-β1 complex from electric eel. Cell 170(3):470–482
Yu FH et al (2003) Sodium channel β4, a new disulfide-linked auxiliary subunit with similarity to beta2. J Neurosci 23(20):7577–7585
Yuan L et al (2014) Investigations of the Navβ1b sodium channel subunit in human ventricle; functional characterization of the H162P Brugada syndrome mutant. Am J Phys Heart Circ Phys 306(8):H1204–H1212
Zhang MM et al (2013) Co-expression of Navβ subunits alters the kinetics of inhibition of voltage-gated sodium channels by pore-blocking μ-conotoxins. Br J Pharmacol 168(7):1597–1610
Zhao J, O'Leary ME, Chahine M (2011) Regulation of Nav1.6 and Nav1.8 peripheral nerve Na+ channels by auxiliary β-subunits. J Neurophysiol 106(2):608–619
Zhou T-t et al (2012) Glycosylation of the sodium channel β4 subunit is developmentally regulated and involves in neuritic degeneration. Int J Biol Sci 8(5):630
Zhu W et al (2017) Mechanisms of noncovalent β subunit regulation of Nav channel gating. J Gen Physiol 149:813–831
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter

Cite this chapter
Molinarolo, S., Granata, D., Carnevale, V., Ahern, C.A. (2017). Mining Protein Evolution for Insights into Mechanisms of Voltage-Dependent Sodium Channel Auxiliary Subunits. In: Chahine, M. (eds) Voltage-gated Sodium Channels: Structure, Function and Channelopathies. Handbook of Experimental Pharmacology, vol 246. Springer, Cham. https://doi.org/10.1007/164_2017_75
Download citation
DOI: https://doi.org/10.1007/164_2017_75
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-90283-8
Online ISBN: 978-3-319-90284-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)