
Abstract
Bacterial pathogens interact with various types of tissues to promote infection. Because it controls the formation of membrane extensions, adhesive processes, or the junction integrity, the actin cytoskeleton is a key target of pathogens during infection. We will highlight common and specific functions of the actin cytoskeleton during bacterial infections, by first reviewing the mechanisms of intracellular motility of invasive Shigella, Listeria, and Rickettsia. Through the models of EPEC/EHEC, Shigella, Salmonella, and Chlamydia spp., we will illustrate various strategies of diversion of actin cytoskeletal processes used by these bacteria to colonize or breach epithelial/endothelial barriers.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Actin-based motility
- Chlamydia
- Cytoskeleton
- EPEC/EHEC
- Invasion
- Listeria
- Rickettsia
- Salmonella
- Shigella
- Type III effectors
1 Introduction: Cytoskeletal Remodeling During Bacterial Infections
The ability of the pathogens to promote disease depends critically on their ability to adhere to the target cells. For this, bacteria have developed a variety of adhesins, surface appendages such as fimbriae or pili, often acting as lectins, binding to sugars that are present at the intestinal mucosal surface of the host cells. For these extracellular pathogens, the disease symptoms may be associated with the secretion of toxins or cytotoxins, leading to the destabilization of tissue integrity and loss of homeostasis (Aktories et al. 2011; Lemichez and Aktories 2013). In some instances, however, the loss of tissue homeostasis is not linked to the production of secreted toxins, but to the pathogen’s ability to reorganize the actin cytoskeleton of host cells.
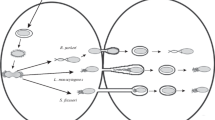
Invasive bacterial pathogens have evolved a diversity of specialized secretion systems allowing the specific injection of effectors into target cells which, as opposed to diffusely acting toxins, function at the close to precisely divert or reprogram processes of the infected cell. The pathogenic bacteria discussed here are Gram negative and express the T3SS protein complex that serves to inject effectors into host cells (for details, see below). For all infectious processes, one can distinguish three phases: at the onset of infection, bacterial pathogens promote their adherence or internalization beyond epithelial or endothelial barriers (Fig. 1, 1). Adhesins or invasins expressed by these bacterial pathogens may show specificities for receptors reflecting tissue tropism. In a second phase and depending on the pathological strategies, disease progression depends on the bacterial ability to multiply within the epithelial/endothelial tissue, to spread from cell to cell using actin-based motility, or to disseminate to deeper tissues (Fig. 1, 2). Specifically for pathogens responsible for chronic infection, persistence may be determined by the bacterial ability to survive within macrophages and reach deeper tissues. Finally, acute phases are associated with the loss of tissue integrity corresponding to the pathogen’s replicative niche, associated with uncontrolled bacterial replication and inflammation (Fig. 1, 3). These phases can be accompanied with the release of bacteria from lysed infected cells, the shedding of bacteria-loaded cells, or cell buds from infected tissues (Fig. 1, 3). In this chapter, we will mostly focus on the initial phases of bacterial infection corresponding to the interactions with epithelial or endothelial tissues.

Colonization and breaching of epithelial/endothelial tissues by T3SS-expressing pathogens. Epithelial/endothelial interaction models of various T3SS-expressing bacteria are depicted. Three stages implicating cytoskeletal reorganization can be distinguished: (1) an initial interaction stage, leading to the injection of T3 effectors inducing adhesion and pedestal formation for EPEC/EHEC, or invasion for Shigella, Salmonella, and Chlamydia (arrow). Following invasion, Shigella rapidly lyses the phagocytic vacuole, while Salmonella and Chlamydia reside and multiply within vacuoles. For Chlamydia, invasion and intracellular replication occur through the EB (small blue circle) and RB (bigger blue circle) forms, respectively; (2) a replication/dissemination stage, where EPEC may surf at the surface of infected epithelial cells, Shigella uses actin-based motility to spread from cell to cell, and Salmonella may infect macrophages and disseminate to other tissues; (3) an acute phase, where bacterial replication is not controlled by host responses, leading to loss of tissue integrity. This stage is usually associated with mounting inflammation and in the case of invasive bacteria, release of bacteria in the extracellular milieu. Such release may correspond to lysis of infected cells or an egress strategy in the case of cells infected with bacteria that freely replicate in the cytosol for Salmonella or the budding of Chlamydia-containing vesicles
2 Bacterial Actin-Based Motility (ABM)
2.1 The ABM Input on Fundamental Principles of Actin Dynamics
A number of invasive bacterial pathogens such as Shigella sp., Listeria monocytogenes, Burkholderia pseudomallei, Mycobacterium marinum, and Rickettsia sp. induce actin comet tails to move intracellularly and spread from cell to cell (Kuehl et al. 2015; Stevens et al. 2006; Truong et al. 2014; Welch and Way 2013; Willcocks et al. 2016).
Actin-based motility of intracellular pathogens has provided key insights into the mechanism regulating actin polymerization. Intracellular motile bacteria were initially observed as inducing the polymerization of long actin comet tails associated with the spreading in epithelial or endothelial cells (Kuehl et al. 2015; Welch and Way 2013). Intracellular motility is promoted by the bacterial surface proteins IcsA/VirG for Shigella and ActA for Listeria (Bernardini et al. 1989; Kocks et al. 1992), the latter being key to the identification of the first cellular endogenous actin nucleator, the Arp2/3 complex from platelet extracts (Welch et al. 1997). ActA and IcsA, localized at one bacterial pole, induce the formation of actin comet tails by directly or indirectly activating the Arp2/3 complex, respectively (Egile et al. 1999; Suzuki et al. 1998; Welch et al. 1998).
Major advances were subsequently achieved using purified proteins to reconstitute bacterial motility in vitro (Loisel et al. 1999). ActA-mediated actin comet tail formation was shown to only require at minima, the Arp2/3-mediated nucleation of filaments, ADF/cofilin, and capping proteins to maintain a high pool of actin monomers at steady state (Loisel et al. 1999). Profilin and the actin-bundling protein alpha-actinin render bacterial motility more effective. (1) ADF/cofilin enhances the depolymerization rate of actin filaments, resulting in an increase in the pool of monomeric ADP-G-actin; (2) profilin binding to ADP-G-actin accelerates nucleotide exchange to regenerate ATP-G-actin and catalyzes its exclusive assembly at barbed ends; (3) by preventing barbed end growth, capping proteins funnel the polymerization flux to newly created uncapped filaments and increase the Arp2/3 branching frequency through the capping of newly created filaments (Akin and Mullins 2008; Pantaloni et al. 2001; Wiesner et al. 2003). Actin filaments polymerizing against the bacteria create a propelling pushing force (Loisel et al. 1999; Theriot et al. 1994) that has been modeled at different scales. At the molecular scale, in the “elastic Brownian ratchet” model, the incorporation of monomers at the filament’s end is rendered possible by the undulations of the actin fiber, the growing filament providing an elastic force pushing the bacteria forward (Mogilner and Oster 1996, 2003). At the mesoscopic scale, the actin meshwork growing from the bacterial surface is considered as a viscoelastic gel, responsible for elastic forces in addition to those mediated by the pushing actin comet tail and accounting for the saltatory intracellular motility observed for some bacteria (Boukellal et al. 2004; Gerbal et al. 2000; Jasnin et al. 2013; Soo and Theriot 2005).
2.2 Bacterial Models of Actin-Based Motility
Shigella flexneri and S. dysenteriae, the causative agents of bacillary dysentery, invade the colonic mucosa where they elicit an intense inflammation responsible for the tissue destruction. Following uptake by intestinal epithelial cells, Shigella lyses the phagocytic vacuole to replicate in the cytosol. During this replication phase, Shigella uses actin-based motility to spread from cell to cell (Fig. 2). Shigella also uses a T3SS to invade and to disseminate within intestinal epithelial cells. To activate Arp2/3, the Shigella IcsA/VirG protein mimics the activated form of Cdc42 by binding to the N-terminal autoregulatory domain of the N-WASP NPF (Fig. 2a). This interaction releases the C-terminal VCA domain of N-WASP, which binds to and activates Arp2/3 (Egile et al. 1999; Rohatgi et al. 1999; Suzuki et al. 1998, 2002). In addition, the Btk and Abl kinases that phosphorylate N-WASP (Burton et al. 2005; Dragoi et al. 2013), as well as Toca-1, which prevents N-WASP inhibition by WIP, are also major components required for Shigella actin-based motility (Ho et al. 2004; Leung et al. 2008).

Bacterial actin-based motility and intercellular spreading. (a) Bacterial actin-based motility. The Shigella autotransporter surface protein IcsA binds to N-WASP and activates Arp2/3-dependent actin polymerization to generate an actin comet tail composed of branched filaments. T3SS also triggers the recruitment of Toca-1 that relieves the WIP-mediated inhibition of N-WASP. The Rickettsia uses two modes of actin-based motility via distinct bacterial surface proteins: (1) RickA directly binds to Arp2/3 to induce actin polymerization; (2) at later stages of infection, Sca2 induces the polymerization of unbranched actin filaments, in a formin-like manner. The Listeria ActA protein activates Arp2/3 and binds to VASP. Lamellipodin (Lpd) is also recruited at the bacterial surface to enhance VASP-dependent bacterial motility and directionality. (b) The formation of Shigella protrusion is favored by the formin Dia1 and by tricellulin at multi-junctions (1). Protrusion elongation is facilitated by Myosin-X. PI(3,4,5)P3 at the protrusion membrane may trigger the activation of PI3K and the phagocytosis of the protrusion by the recipient cell in a noncanonical clathrin and dynamin-dependent pathway (2). Listeria-containing protrusion formation is favored by the formins Dia1, Dia2, and Dia3. The InlC protein releases cell-cell junctions’ tension by interfering with tuba-mediated activation of N-WASP, thus favoring protrusion formation. Tuba contributes to cell-cell junctions’ tension trough its GEF activity for Cdc42 or by allowing the recruitment of N-WASP at cell junctions. The tuba GEF (blue inset) and SH3 (pale green) domains are depicted. An unidentified Listeria factor (?) also contributes to cortical tension release by inhibiting Cdc42
Rickettsia spp. are obligate intracellular bacteria that include R. parkeri, R. rickettsii, and R. conorii, the causative agents of spotted fever. Rickettsia also uses actin-based motility to disseminate within tissues. Rickettsia-mediated actin comet tails consist of long and parallel actin filaments (Gouin et al. 1999; Van Kirk et al. 2000). The discovery of RickA, a surface protein of Rickettsia that acts as a NPF for Arp2/3 through a canonical VCA domain, was puzzling, since this protein induced a branched actin network (Gouin et al. 1999; Harlander et al. 2003; Jeng et al. 2004). However, recent studies showed that Rickettsia actin-based motility involves two distinct phases (Fig. 2a). The first phase based on RickA activity occurs within the first 2 h following bacterial internalization, during which short Arp2/3-containing actin comet tails propel bacteria in the cytosol at a slow speed. The second phase occurring after 12–24 h post internalization is based on Sca2, another Rickettsia surface protein (Reed et al. 2014). Sca2 nucleates unbranched actin filaments while remaining attached to the barbed end of elongating actin filaments, thus accelerating actin assembly in a formin-like manner, but using a different molecular mechanism (Haglund et al. 2010; Reed et al. 2014) (Fig. 2a). This formin-like nucleation and accelerated processive elongation of actin filaments explain the structure of actin comet tails associated with bacteria and the enhanced velocity of bacterial propulsion (Gouin et al. 1999; Reed et al. 2014).
Listeria monocytogenes is an enteroinvasive bacterium responsible for septicemia, encephalomeningitis, and abortion. Following ingestion and invasion of the intestinal mucosa, Listeria uses actin-based motility to disseminate within infected tissues. The Listeria ActA protein acts as a NPF, which, through a VCA domain located at its N-terminal part, directly activates Arp2/3 (Egile et al. 1999; Welch et al. 1998). In addition to Arp2/3, profilin and VASP have been involved in the formation of Listeria actin comet tails (Smith et al. 2010) (Fig. 2a). The proline-rich domain of ActA interacts with the EVH1 domain of VASP, and profilin is recruited through the proline-rich domain of VASP (Niebuhr et al. 1997; Reinhard et al. 1995). VASP tetramers in cooperation with profilin can processively accelerate barbed end elongation in a formin-like manner (Breitsprecher et al. 2011). Thus, ActA may nucleate new actin filaments through Arp2/3 activation and processively elongate these filaments by recruiting the VASP/profilin (Fig. 2a). This model is consistent with the role of VASP in the shape of Listeria actin comet tails and the increased bacterial velocity, as well as in the elastic modulus of the actin comet tails (Samarin et al. 2003; Auerbuch et al. 2003; Diakonova et al. 2007; Suei et al. 2011). Lamellipodin, a cytoskeletal protein involved in cell migration, is also recruited at Listeria surface, via its Ena-VASP homology 1 (EVH1) and plekstrin homology (PH) domains interacting, respectively, with VASP and PI(3,4)P2 present at the surface of intracellular bacteria in an ActA-dependent manner (Wang et al. 2015). As for VASP depletion, depletion of Lamellipodin also affects Listeria intracellular velocity and directionality, suggesting a role for Lamellipodin as a scaffold protein organizing ActA and VASP at the surface of bacteria.
Propulsion of bacteria against the plasma membrane induces filopodia-like protrusions that are internalized by the neighboring cells allowing bacterial spreading. However, recent studies showed that other cellular factors facilitate the formation of bacteria-containing protrusions. For Shigella, the formin nucleator Dia might contribute to force generation required to form protrusion at cell-cell junctions (Heindl et al. 2010). Myosin-X could also contribute to protrusion formation by carrying membranes or host cell components at the protrusions’ tip (Bishai et al. 2013) (Fig. 2b). Phagocytosis of the protrusions also requires active signaling to neighboring cells, such as a clathrin noncanonical pathway implicating the multicellular junction protein tricellulin, PI3K, and dynamin and myosin II (Fukumatsu et al. 2012; Lum and Morona 2014; Rathman et al. 2000) (Fig. 2b).
In the case of Listeria, the 3 Dia formins may also play a similar role in facilitating protrusion formation (Fattouh et al. 2015). In addition, the high tension at cell-cell contacts was shown to limit the formation of protrusions and cell-to-cell spread (Fig. 2b). By acting on tuba, an activator of N-WASP and GEF for Cdc42, the Listeria-secreted protein InlC releases junctional tension (Rajabian et al. 2009). Clearly, beyond actin-based motility, much is yet to be learned about cytoskeletal processes that involved bacterial dissemination in tissues.
3 Induction and Hijacking of Filopodia by EPEC/EHEC and Shigella
Filopodia are fingerlike thin cell extensions present on the surface of a variety of cell types, including epithelial and endothelial cells. Filopodia are generally considered as sensing organelles that cells use to probe their environment to establish adhesion structures. An increasing number of viral and bacterial pathogens such as HIV, or MLV, Yersinia, Shigella, or Borrelia have been reported to divert filopodial function to adhere to or invade host cells (Hoffmann et al. 2014; Lehmann et al. 2005; Romero et al. 2011; Young et al. 1992).
Enteropathogenic E. coli (EPEC) is an important cause of infantile diarrhea in developing countries. Upon interaction with enterocytes, EPEC induces the disappearance of microvilli in a process referred to attaching-effacing (A/E) lesions. The A/E lesions induced by EPEC/EHEC strains are closely linked to virulence, with the loss of electrolytes from infected enterocytes and epithelial integrity. EPEC/EHEC A/E lesions are associated with the formation of F-actin-rich structures supporting bacteria coined “pedestals” (Fig. 3a). The physiological relevance of EPEC/EHEC pedestals has been a matter of debate.

Immunofluorescence micrographs of EPEC- and Shigella-induced cytoskeletal reorganization at the surface of epithelial cells. HeLa cells were infected for 45 min (a) or 15 min (b) with bacteria. Samples were fixed and processed for F-actin (green) and bacterial (red) staining. Various F-actin-containing structures are observed. EPEC induces the formation of pedestals composed of several actin shafts, reminiscent of the “medusa” phenotype (a, left; (Smith et al. 2010)) and pedestal-like structures (a, right). Shigella induces the formation of actin foci-forming filopodia (b, left) and ruffles (b, right). Scale bar = 5 μm
During early phase of adhesion, EPEC and EHEC induce filopodia-like structures at the sites of bacterial contact with epithelial cells (Kenny et al. 2002). These filopodia only form transiently and disappear as actin pedestals are formed. EPEC-/EHEC-induced filopodia are triggered by the type III effector MAP, acting as a GEF for the Cdc42 GTPase (Huang et al. 2009). In addition to its GEF domain, MAP contains a carboxyterminal PDZ (PSD-95/Disc Large/ZO-1) domain that interacts with the ezrin-binding protein Epb50. Binding of MAP to Ebp50 and ezrin promotes the formation of MAP scaffolds at the plasma membrane, which determine the local activation of Cdc42 and actin polymerization (Alto et al. 2006). The role of MAP and filopodia during EPEC/EHEC infection remains unclear. MAP was shown to synergize with the Tir-translocated effector to promote EPEC/EHEC invasion of epithelial cells (Jepson et al. 2003). An EPEC mutant lacking MAP induces the formation of pedestal structures, which do not appear to support the stable adhesion of bacteria (Shaw et al. 2005). This evidence suggests that, rather than performing antagonistic functions, MAP and other type III effectors contribute to the “maturation” of pedestals through the activation of Cdc42 and actin polymerization. MAP-induced filopodia may increase the surface of contact between bacteria and the plasma membrane, thereby stabilizing EPEC/EHEC adhesion upon pedestal formation.
To invade host cells, viruses glide along the side of filopodia in a myosin-II-dependent process. In contrast, prior to invasion of epithelial cells, Shigella interacts with the tip of filopodia, presumably via the T3SS tip complex. Following contact, filopodia retract and drag bound bacteria into contact with the cell body (Fig. 3b). This retraction process depends on the activity of MAP kinase Erk, which controls the retrograde flow of actin filament bundles in filopodia in epithelial cells (Romero et al. 2011). This retrograde flow is driven by the polymerization of actin in the cortical actin network that constantly pulls on filopodial actin filaments. Upon bacterial interaction, filopodia retract because actin polymerization at filopodia tips fails to compensate the pulling of filopodia actin filaments by the retrograde flow (Bornschlogl et al. 2013). The mechanism underlying the decrease of actin polymerization at the filopodia tip following bacterial contact has not been elucidated, but is likely to involve cell receptors of the T3SS tip complex and barbed end capping proteins. Filopodial capture may represent an explanation for the paradoxical cell invasion properties of Shigella in the absence of constitutive cell-binding activity.
4 Intracellular Delivery of Bacterial Effectors via “Injectisomes”
To date, up to nine types of secretion systems have been described in bacteria, three of which enable the delivery of effectors into host target cells, illustrating the striking capacity of pathogens to adapt and improve molecular machines. Historically the type III secretion system (T3SS) was the first one described to allow the injection of bacterial effectors into host cells. T3SSs are widely spread among Gram-negative bacterial pathogens and are expressed in a wide variety of enteropathogens, such as EPEC/EHEC, Salmonella enterica, Yersinia, or Shigella spp. The structure of these macromolecular machines is related to the bacterial flagella, with the notable difference that the basal body is prolonged by a needle and a so-called tip complex. This tip complex negatively regulates secretion and allows sensing of host cell membranes (for review see Galan et al. 2014). While there may be variations between bacterial models, it is generally admitted that the mechanism of type III secretion is conserved. Upon host cell contact, the T3SS activity is triggered, leading to the insertion of two hydrophobic proteins, called the translocon, into the host cell plasma membrane. The translocon forms a ring connecting to the T3SS needle through which type III effectors are channeled directly into the host cell cytosol without extracellular steps. In the case of Yersinia, however, it has been shown that type III substrates can also follow an alternate, AB5-toxin-like route of delivery into host cells (Akopyan et al. 2011). Because of the estimated inner diameter of the T3SS needle or the T4SS secretion conduit (1.5–3 nm), it is thought that substrates are secreted in a fully or partially unfolded state, to adopt their final conformation upon translocation into host cells. These considerations are important, because post-secretion folding may potentially provide means to control the effector activity, for example, by posttranslational modification or association with host cell cofactors (Cui and Shao 2011).
4.1 WXXXE Effectors
A variety of EPEC, Shigella, and Salmonella type III effectors contain the “WXXXE” sequence (Orchard and Alto 2012). Structural and in vitro assays confirmed that WXXXE effectors are GEFs for Rho GTPases. Strikingly, structural analysis of WXXXE effectors revealed a highly conserved fold, with two helical bundles forming a V-shaped structure connected by a “catalytic” loop. The conservation of the WXXXE motif, located at the interface between the two bundles, is explained by its key structural role. All WXXXE effectors promote their GEF activity by interacting via key residues located in their alpha-2 helix and catalytic loop, with the Rho GTPase Switch1 and Switch2 domains (Huang et al. 2009). Sequence comparisons, supported by modeling and mutagenesis studies, indicate that the specificity of interaction is determined by contacts between residues of the alpha-2 and alpha-6 helixes of the WXXXE effector that pair to the beta-2–3 inter-switch strand residues of the GTPase (Huang et al. 2009).
5 Cytoskeletal Remodeling at the Plasma Membrane by T3SS-Expressing Bacteria
5.1 EPEC/EHEC Pedestals
Through actin treadmilling, EPEC-mediated pedestal formation is associated with the “surfing” motility of bacteria on the cell surface and has been proposed to enable bacterial dissemination over the intestinal mucosa (Lai et al. 2013; Wong et al. 2011) (Fig. 3). Alternatively, EPEC-mediated pedestal formation on epithelial cells (Fig. 3a) may reflect the diversion of cytoskeletal processes involved in the inhibition of phagocytosis by macrophages (Fallman et al. 2002; Westermark et al. 2014).
EPEC pedestal formation is dependent on the injection of effectors mediated by the T3SS. Upon cell contact, EPEC injects the Tir protein that inserts in host cell plasma membrane (Fig. 4). Folding of Tir in the plasma membrane results in the cell surface exposure of a loop, which is recognized by the EPEC/EHEC intimin, a bacterial surface adhesin containing a cell-binding domain with structural similarities with the Yersinia invasin protein. As commonly observed during cell outside-in receptor-mediated signaling, clustering of Tir molecules by multimers of intimin induces the tyrosyl phosphorylation of Tyr474 of Tir. Host cell tyrosine kinases involved in Tir phosphorylation include members of the Src, Arg/Abl, or Tec family kinases (Bommarius et al. 2007; Swimm et al. 2004) (Fig. 4). Tir phosphorylation probably involves loops of amplification through the recruitment and activation of various tyrosine kinases. Tir phosphorylation at Y474 mediates the recruitment of the Nck adaptor through its SH2 domain. Nck SH3 domains, in turn, allow the recruitment and activation of N-WASP, itself responsible for the activation of the Arp2/3 complex leading to pedestal formation (Campellone and Leong 2003) (Fig. 4). This view, however, is probably oversimplistic and has been challenged by the observations that N-WASP is required for the recruitment of Nck and that the Nck-binding region of N-WASP is dispensable for its localization at pedestal (Garber et al. 2012). While Tir Y474 phosphorylation is the major route for actin pedestal formation, the existence of Nck-independent pathways involving two other phosphorylated tyrosine residues of Tir could account for N-WASP recruitment in the absence of direct interaction with Nck (Lai et al. 2013).

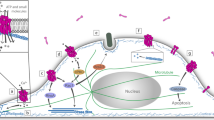
Common and specific processes targeted by T3SS-expressing bacteria to induce cytoskeletal reorganization at the host cell plasma membrane. Components implicated in cytoskeletal reorganization are indicated. Boxes correspond to pale yellow, tyrosine kinases (red), adaptor (orange), phosphoinositides (green), BAR-domain-containing proteins, and GEFs (purple); dark yellow, Rho GTPases; orange, WAVE-family proteins; green, cytoskeletal proteins; and blue circles, type III effectors. Solid arrows indicate direct interaction/activation. Dashed arrows indicate activation. See main text for description. (EPEC/EHEC), gray box: type III effectors contributing to cytoskeletal reorganization but not critical for pedestal formation. F EspF, Fu EspFu/Tccp, H EspH, T EspT, M2 EspM2. (Shigella), A IpaA, B1 IpgB1, B2 IpgB2, D IpgD. (Salmonella), C SipC, B SopB, E SopE, E2 SopE2, A SipA. (Chlamydia), P TarP
In EHEC, tyrosine phosphorylation at Y458 (the EPEC equivalent of Y454) does not appear to play a prominent role in actin polymerization and pedestal formation (Fig. 4). Instead, the EHEC NPY458 motif was shown to bind to the I-BAR containing proteins IRTKS and Irsp53. In addition, EHEC injects the type III effector EspFu/Tccp that binds to the carboxyterminal SH3 domains of IRTKS/Irsp53 through a proline-rich repeated sequence and to N-WASP through an N-terminal helix. The EspFu N-terminal helix acts as a super-mimic that outcompetes the N-WASP inhibitory helix and relieves its intramolecular interaction with the Arp2/3-activating domain VCA (Lai et al. 2013). While this Tir “NPY”dependent actin polymerization pathway appears to function with low efficiency in EPEC, the presence of EspFu/Tccp renders it predominant in EHEC (Campellone and Leong 2003). Other minor pathways include the EPEC Tir-dependent Nck-independent and EHEC EspFu-/Tccp-dependent N-WASP-independent pathways (Wong et al. 2011). Interestingly, some EPEC strains also express EspF, which shares homology and can functionally substitute for EHEC EspFu/Tccp. Through its proline-rich repeats, however, EspF was shown to bind and activate Snx9, a BAR-domain protein, leading to the formation of tubular membrane structures (Alto et al. 2007). The interaction of EspF with Snx9 appears to determine its localization at the plasma membrane and N-WASP-dependent actin polymerization, although its role during bacterial infection is not entirely clear.
While MAP and Tir are present in all EPEC/EHEC strains, other EPEC/EHEC type III effectors that are strain specific might control cytoskeletal reorganization. Because the functions of these effectors are potentially antagonistic, their expression is likely to be coordinated during the course of the infection. The type III effector EspH binds the Dbl homology/pleckstrin homology domains present in endogenous GEFs, thereby inhibiting the GTPase activation (Wong et al. 2012). EspM1/2 is a WXXXE effector acting as a RhoA GEF. Expression of EspM1/2 accelerates the disappearance of pedestals and perturbs the distribution of tight junction components in polarized epithelial cells, while decreasing the epithelial barrier permeability (Simovitch et al. 2010). The function of EspM1/2 during infection is not clear; as postulated for the CNF1/2 toxins, EspM1/2 may serve to limit inflammation associated with leakage from tight junctions (Arbeloa et al. 2010). EspT, another WXXXE effector expressed only in a minority of EPEC strains, is a GEF for Cdc42 and Rac, associated with membrane ruffling and with higher invasive properties (Bulgin et al. 2009). The EspH inhibitory activity on GTPases has been linked to the ability of EPEC strains to prevent their phagocytosis by macrophages and to inhibition of MAP-induced filopodia. While inhibiting Rho GTPase activation, EspH stimulates actin polymerization and the formation of EPEC pedestals in a Tir-WIP- and N-WASP-dependent but Nck- and Rac-independent manner. These seemingly paradoxical functions of EspH have led to speculations that EPEC/EHEC strains “reprogram” signaling downstream of Rho GTPases (Cui and Shao 2011). In this view, “reprogrammed” cytoskeletal reorganization would ensue from the capacity of EspH to inhibit the activation of endogenous Rho GTPases, without affecting that of WXXXE effectors (Cui and Shao 2011).
5.2 Shigella Invasion
Upon host cell contact, activation of the Shigella T3SS leads to the insertion of the IpaB and IpaC translocon components, required for the injection of type III effectors (Picking and Picking 2016). In addition to being involved in type III effector translocation, IpaC also triggers the recruitment and activation of the Src tyrosine kinase through its carboxyterminal moiety, by a process that remains ill defined (Fig. 4). Src-dependent tyrosyl phosphorylation of cortactin allows its interaction with the Crk adaptor at the plasma membrane and actin polymerization via the cortactin Arp2/3-binding domain (Valencia-Gallardo et al. 2015). Inhibition of Src- and cortactin-mediated signaling results in actin-rich smaller structures and in a partial defect in bacterial internalization, suggesting that rather than triggering, Src signaling amplifies actin polymerization at bacterial invasion sites (Bougneres et al. 2004). Cytoskeletal reorganization also results from the activity of injected type III effectors acting at different levels to control dynamics of actin polymerization/depolymerization. IpgB1 and IpgB2 are WXXXE effectors acting as GEFs for Rac and Rho, respectively (Alto et al. 2006). While the activation of Rac and Rho is generally considered antagonistic, both IpgB1 and IpgB2 are required for efficient Shigella invasion in polarized intestinal epithelial cells for reasons that are still unclear (Hachani et al. 2008). Through its GEF activity for Rac, IpgB1 stimulates actin polymerization and membrane ruffling at invasion sites, thereby amplifying Shigella invasion. IpgB1 has also been involved in the hijacking of the ELMO/Dock180 pathway, by acting as a mimic of the RhoG GTPase (Handa et al. 2007). In light of compelling evidence for the role of IpgB1 as a Rac GEF, how this WXXXE effector could also mimic the function of RhoG deserves clarification. Interestingly, other studies have shown that Arg/Abl are involved in actin polymerization at Shigella invasion sites and in bacterial entry (Burton et al. 2003; Wessler and Backert 2011). Abl/Arg are tyrosine kinases implicated in cytoskeletal reorganization downstream of growth hormone receptor signaling. Transactivation of Abl/Arg may occur upon binding of their proline-rich domain to the Crk adaptor, but the activity of these kinases is also regulated by direct interaction with cortactin. Abl/Arg can also act upstream of the Rac GTPase by stabilizing a Crk/ELMO/Dock180 complex (Wessler and Backert 2011). It is therefore likely that actin polymerization during Shigella invasion is subjected to loops of amplification dependent on tyrosine kinase signaling and Rac GTPase, making the deciphering of pathways downstream of Shigella effectors a complex task.
5.3 Salmonella Invasion
Salmonella enterica enterica regroups various serovars predominantly responsible for food-borne gastroenteritis and enteric fever. The vast majority of studies have been performed on Salmonella enterica serovar Typhimurium. Salmonella contains two pathogenicity islands SPI-1 and SPI-2, each encoding a T3SS critical for virulence. While the SPI-2 locus is implicated in intracellular survival in macrophages and bacterial dissemination to various organs, the SPI-1 T3SS is involved in the crossing of the intestinal barrier and cytoskeletal reorganization during Salmonella invasion (Valdez et al. 2009). Interestingly, similar actors involved in tyrosine kinase signaling have also been implicated during Salmonella invasion (Ly and Casanova 2009; Shi and Casanova 2006). Consistent with the notion that T3SSs may bypass receptor-mediated signaling, Salmonella invasion implicates many components involved in cell adhesion, with the exception of beta 1 integrins (Fig. 4). During cell adhesion, tyrosyl phosphorylation of the focal adhesion kinase FAK determines the recruitment of the Src kinase and subsequent maturation of adhesion structures through the scaffolding activity of adaptor proteins such as p130Cas (Shi and Casanova 2006) (Fig. 4). FAK and p130Cas are also involved in Salmonella invasion, in a process that does appear to depend on the FAK kinase but through its scaffolding activity. Although their inactivation leads to a reduction of bacterial-induced actin foci of invasion, FAK and Cas play distinct roles during Salmonella invasion, since, as opposed to FAK −/− cells, remaining foci in Cas −/− cells also present a defect in phagocytic cup formation suggesting a defect in actin organization (Shi and Casanova 2006). Also, Salmonella invasion does not require the focal adhesion protein paxillin, suggesting that bacterial invasion implicates different levels of cytoskeletal tethering than those associated with the maturation of cell adhesion structures. Like for Shigella, the CrkII adaptor, as well as the Arg/Abl kinases, participates in Salmonella invasion suggesting common invasion mechanisms between these bacterial pathogens (Ly and Casanova 2009).
The type III Salmonella effector SopE is a WXXXE effector acting as a GEF for Cdc42 and Rac, implicated in actin polymerization and Arp2/3-dependent membrane ruffling during bacterial invasion (Hardt et al. 1998; Orchard and Alto 2012). SopE activates Cdc42 and Rac during bacterial invasion. The role of Cdc42 during invasion, however, requires clarification. Targeted RNAi inhibition indicated that Cdc42 was dispensable and Rac1 was essential for actin polymerization and membrane ruffling at invasion sites (Patel and Galan 2006).
The sopE gene is not present in all pathogenic Salmonella enterica serovars, as opposed to sopE2 sharing 64% identity with sopE, and showing a broader specificity. As opposed to SopE, SopE2 shows GEF activity specific to Cdc42 (Friebel et al. 2001). Inactivation of sopE or sopE2, as opposed to double mutants, leads to partial inhibition of membrane ruffling and bacterial invasion, indicating a joint action of these type III effectors (Zhou et al. 2001). While actin polymerization determines membrane ruffling during the early stages of Salmonella contact with host cells, actin depolymerization is required to complete the internalization process. To this end, Salmonella injects another type III effector, SptP, into host cells, which has a dual tyrosine phosphatase and GAP activity toward Cdc42 and Rac (Stebbins and Galan 2001). The dual activity of SptP depends on two distinct domains: an N-terminal GAP domain and a C-terminal tyrosine phosphatase domain. The SptP GAP domain shares sequence and structural homologies with the Pseudomonas ExoS and Yersinia YopE type III effectors, while the tyrosine phosphatase domain shows some homology with the Yersinia YopH phosphatase (Stebbins and Galan 2001). The SptP GAP domain is the smallest characterized RhoGAP and is structurally distinct from endogenous GAPs. Various structural SptP features argue in favor of convergent evolution, where minimal domains of various eukaryotic GAPs appear to have been hijacked to compose a particularly effective bacterial GAP (Stebbins and Galan 2001). In particular, the catalytic mechanism of SptP is similar to that of endogenous GAPs and involves a so-called arginine finger, with an arginine residue exposed in the GTPase active site and neutralizing the negative charge at the β-phosphate during the GTPase transition state. In the case of SptP, however, the arginine residue is exposed on a helical bundle, as opposed to the loop structure of endogenous “arginine fingers.” The regulation of potentially antagonistic activities of SopE/SopE2 and SptP was proposed to be mediated by a differential stability of these type III effectors following injection into host cells, following their ubiquitination and proteasomal degradation (Kubori and Galan 2003). Imaging of the kinetics of delivery of these effectors into host cells also argues that SopE is injected faster than SptP, providing another levels of temporal control of their activity during invasion (Van Engelenburg and Palmer 2008).
T3SS-mediated invasion by Salmonella was shown to involve two distinct signaling pathways. In addition to the SopE-Rac-/Cdc42-dependent pathway leading to Arp2/3 complex-dependent actin polymerization and membrane ruffle formation, the type III effector SopB induced the activation of the RhoA GTPase leading to Rho kinase (ROCK) activation and bacterial uptake in a myosin-II-dependent process (Hanisch et al. 2011). In independent studies, SopB was shown to mediate the activation of the FHOD-1 formin downstream of ROCKI (Truong et al. 2013). In these aspects, Salmonella invasion presents similarities to CR3-mediated phagocytosis in macrophages, which involves a Rac-WAVE-Arp2/3 and a RhoA-formin-/myosin-II-dependent pathway (Sarantis and Grinstein 2012). However, it is unclear how independent the SopE- and SopB-mediated pathways are, since SopE is also involved in FHOD-1 phosphorylation, and as described earlier, SopB synergizes with SopE to mediate activation of the Abi1-WAVE complex and Arp2/3-dependent actin polymerization (Humphreys et al. 2012; Truong et al. 2013).
5.4 Chlamydia Invasion
Chlamydia trachomatis is a member of the obligate intracellular bacterial pathogens Chlamydiae, responsible for infectious keratoconjunctivitis, a predominant cause of blindness in developing countries, and for sexually transmitted diseases, which can lead to infertility (Bastidas et al. 2013; Mehlitz and Rudel 2013). Chlamydia has a biphasic cycle with elementary bodies (EBs) able to resist in the extracellular environment and invade host cells and reticulate bodies (RBs) associated with intracellular replication (Fig. 1). While the genetic manipulation of Chlamydiae is still at its infancy, the genomic analysis and use of orthologous systems have allowed significant advance in the study of bacterial invasion mechanisms. Various adhesins triggering host cell signaling during initial interactions with the host have been described and reviewed elsewhere (Mehlitz and Rudel 2013). Through screening using RNA interference, tyrosine kinase signaling implicating the Arg/Abl family was shown to be required for C. trachomatis invasion (Elwell et al. 2008; Mital and Hackstadt 2011). Remarkably, Chlamydia also possesses a T3SS that has been involved in the cytoskeletal reorganization during invasion, in particular, through the injection of the T3 effector TarP (Clifton et al. 2005) (Fig. 4). TarP is phosphorylated by Arg/Abl and Src kinases at the level of tyrosine-rich tandem repeats triggering the recruitment of the Rac GEFs Sos1 and Vav2 (Jewett et al. 2008; Lane et al. 2008). Phosphopeptide pulldown experiments showed that Sos1 associates with the TarP tyrosyl-phosphorylated domain through Abi1 and Eps8. Vav2, on the other hand, requires PI(3,4,5)P3 synthesized by class I PI3Ks also recruited at bacterial cell interaction sites, through its interaction with the TarP phosphorylated domain (Lane et al. 2008). Thus, following Arg/Abl-mediated tyrosyl phosphorylation, TarP permits the recruitment of the Sos1 and Vav2 GEFs, Rac, and the Abi1-WAVE2. The recruitment of the Abi1-WAVE2 complex downstream of Rac activation is potentiated by the concurrent activation of Arf6 and Arf1 to trigger Arp2/3 complex-dependent actin polymerization (Krause and Gautreau 2014). While the precise link with TarP-mediated activation of Rac has not been established, former studies have shown that Arf6 is required for Chlamydia-mediated cytoskeletal rearrangements suggesting the induction of pathways similar to those observed during Shigella and Salmonella invasion (Balana et al. 2005). Downstream of the tyrosine-rich domain, TarP contains an LD motif similar to that found in the focal adhesion protein paxillin, involved in the recruitment of the focal adhesion kinase (FAK) (Thwaites et al. 2014). TarP-mediated FAK recruitment is required for the downstream activation of Cdc42 and Arp2/3 complex-dependent actin polymerization. The molecular links between FAK and Cdc42 leading to the activation of the GTPase and actin polymerization during Chlamydia invasion are not as yet identified and could potentially include the GEFs from the DOCK family (Thwaites et al. 2014). Hence, through various modules, TarP may trigger actin polymerization through tyrosine kinase signaling and activation of the Cdc42 and Rac GTPases.
6 The Role of Phosphoinositides During T3SS-Mediated Cytoskeletal Reorganization
Phosphatidyl inositol phosphates (PIPs) are implicated in fundamental cell processes, including vesicular trafficking and cytoskeletal remodeling (Sarantis and Grinstein 2012; Viaud et al. 2016). The phosphatidyl moiety of PIPs mediates their association with membranes, and various kinases and phosphatases phosphorylate/dephosphorylate the inositol ring at different positions, generating a variety of second messengers (Viaud et al. 2016). Among these, PI(4,5)P2 regulates the localization of cytoskeletal linkers at the plasma membrane by allowing their recruitment through so-called pleckstrin homology (PH) domains. PI(3,4,5)P3 is upregulated following activation of class I PI3Ks downstream of receptor-mediated signaling and is pivotal in the recruitment of GEFs for Rho GTPases or actin nucleating-promoting factors (Viaud et al. 2016).
The Shigella type III effector IpgD was found to harbor a PI(4,5)P2-5 phosphatase activity leading to the synthesis of PI5P (Niebuhr et al. 2002). IpgD has been implicated in various processes during Shigella infection, which are linked to PI(4,5)P2 hydrolysis or PI(5)P synthesis. Through its PI(4,5)P2 hydrolysis, IpgD was shown to disconnect cortical actin from the plasma membrane as shown by the membrane-pulling experiments using beads manipulated by magnetic tweezers in IpgD-transfected cells (Niebuhr et al. 2002). In epithelial cells, disconnection of cortical actin by IpgD would facilitate actin polymerization at bacterial invasion sites. In T lymphocytes, however, hydrolysis of P(4,5)P2 at the plasma membrane by injected IpgD prevents cell migration via the phosphorylation of ERM proteins, thereby impeding cell migration and function (Konradt et al. 2011). These results indicate that through PI(4,5)P2 hydrolysis, Shigella not only locally regulates the dynamics of actin polymerization but also may interfere with the global cytoskeletal machinery. PI(5)P synthesized by IpgD may also directly bind to the PH domain of Tiam 1 and stimulate its exchange factor for Cdc42 and Rac (Viaud et al. 2014). While activation of Tiam 1 leading to Rac-dependent actin polymerization and ruffle formation was demonstrated in cells transfected with IpgD, it remains unclear whether this pathway is triggered during Shigella invasion. IpgD upregulates the levels of PI(3,4,5)P3 at entry sites, presumably through an interplay between PIPs phosphatases and kinases, and the activation of PI3K (Garza-Mayers et al. 2015). PI3K has been involved in the regulation of Rac activity through the activation of various GEFs. In the case of Shigella, PI3K activation is required for the recruitment of ARN0, a GEF for the Arf6, a GTPase involved in endocytic process at the plasma membrane and endosome recycling (Garza-Mayers et al. 2015). Arf6 has also been implicated in the activation of the Rac GEF ELMO/Dock180 (Santy et al. 2005). Because Arf6 also contributes to actin polymerization at Shigella invasion sites in an IpgD-dependent manner, it is proposed that IpgD triggers a positive feedback loop implicating PI3K-ARNO-Arf6 that amplifies Rac-dependent actin polymerization (Garza-Mayers et al. 2015).
Interestingly, prior to the Shigella study, it was shown that the Salmonella type III effector SopB triggers a similar pathway involved in actin polymerization during bacterial invasion (Humphreys et al. 2012). SopB was first characterized as an inositol-polyphosphate (InsPPs) phosphatase leading to the production of InsP6, involved in Salmonella invasion of epithelial cells (Zhou et al. 2001). It became clear that SopB also harbored PIPs dual 4- and 5-phosphatase activity and that both activities were required for bacterial-induced cytoskeletal remodeling (Piscatelli et al. 2016). SopB leads to higher levels of PI(3)P, either through its direct 4-phosphatase activity toward PI(3,4)P2 or via the Rab5- and Vps34-dependent recruitment of PI(3)P-enriched vesicles, at Salmonella invasion sites (Mallo et al. 2008). In a process that is not fully understood, SopB is also responsible for the increases in PI(3,4)P2 and PI(3,4,5)P3 at Salmonella invasion sites in a wortmannin-insensitive manner, suggesting that it is independent of PI3K (Mallo et al. 2008).
In response to the PI(3,4,5)P3 increase induced by SopB, ARNO was shown to recruit and activate Arf6 (Humphreys et al. 2012). It is not known, however, whether Arf6 recruitment stimulates the activation of a Rac GEF such as DOCK/ELMO180 during Salmonella invasion. Instead, Arf6 leads to the recruitment of Arf1, a GTPase involved in the trans-Golgi network trafficking. Arf1, in turn, is proposed to stimulate the activation of the Abi/WAVE complex downstream of Rac and actin polymerization at Salmonella invasion sites, in a scheme similar to that proposed for lamellipodia formation upon growth factor receptor stimulation (Humphreys et al. 2012; Krause and Gautreau 2014). Consistent with a cooperative action of injected type III effectors, Salmonella sopB and sopE double mutants are severely impaired in bacterial invasion (Zhou et al. 2001).
PIPs also regulate EPEC pedestal formation. PI(4,5)P2 is enriched in microdomains at the site of interaction of EPEC/EHEC with the host cell plasma membranes (Sason et al. 2009). Tyrosine phosphorylation of Tir at Y454 was proposed to mediate the recruitment and activation of PI3K through its SH2 domain, which phosphorylates PI(4,5)P2 to generate PI(3,4,5)P3 proposed to participate in actin polymerization during the formation of bacterial pedestals (Sason et al. 2009). How the recruitment of IRTKS/Irsp53 and that of PI3K are regulated by EPEC Tir Y454 is not known. Tir also contains tyrosine-based motifs at its C terminus, similar to ITIM motifs found in immunoreceptors, involved in the recruitment of lipid phosphatases via their SH2 domains (Smith et al. 2010). These motifs, including EPEC Tir Y483 and Y511, serve as docking motifs for the SHIP2 phosphatase that hydrolyzes PI(3,4,5)P3 into PI(3,4)P2. PI(3,4)P2 was shown to permit the recruitment at EPEC pedestal of Lamellipodin through its PH domain, an adaptor protein regulating the dynamics of lamellipodia formation (Smith et al. 2010). SHIP2 was also shown to act as a scaffold by recruiting the cytoskeletal adaptor SHC. Strikingly, upon inhibition of SHIP2, EPEC induces the formation of longer pedestal containing multiple stalks reminiscent of “medusa” tentacles (Fig. 3a) (Smith et al. 2010). Thus, while PI3K kinase, through PI(3,4,5)P3, stimulates Tir-mediated actin polymerization and pedestal elongation, the recruitment of SHIP2, and of Lamellipodin through the synthesis of PI(3,4)P2, is required for the formation of a dense actin network in EPEC pedestals. Interestingly, despite the presence of putative ITIMs motifs in EHEC Tir, SHIP2 does not appear to control the dynamics of EHEC pedestals (Smith et al. 2010). It is possible that the role of SHIP2 in the regulation of actin polymerization in pedestal is restricted to signaling implicating the tyrosyl phosphorylation of Tir Y474 and Nck, as observed for EPEC but not EHEC.
7 Type III Effectors as Actin-Binding Proteins and Cytoskeletal Linkers
Bacterial pathogens may also inject effectors into host cells, which directly affect the dynamics of actin polymerization/depolymerization, or cytoskeletal anchorage. In addition to injected effectors, the Salmonella translocon component SipC was shown to directly nucleate actin polymerization (Hayward and Koronakis 1999). During Salmonella invasion, SipC may contribute to actin polymerization induced by the injected SopE and SopB type III effectors. The Salmonella type III effector SipA was shown to bind to and stabilize actin filaments, acting in concert with SipC to promote Salmonella invasion (McGhie et al. 2001). The crystal structure of the SipA carboxyterminal fourth region shows a packed globular structure with a large basic patch likely involved in the positioning of SipA onto actin filaments, extended at opposite ends by non-globular arms that “tether” opposite actin strands (Lilic et al. 2003). By stabilizing actin filaments, SipA decreases the critical concentration of G-actin monomers, thereby enhancing actin polymerization (Lilic et al. 2003). SipA was also shown to protect actin filaments from disassembly mediated by ADF/cofilin and gelsolin. Both activities, together with the decrease in critical concentration, can account for increased actin polymerization and membrane ruffling at Salmonella invasion sites (Lilic et al. 2003). Remarkably, following bacterial invasion, SipA remains associated with the cytoplasmic side of the Salmonella-containing vacuole (SCV) (Brawn et al. 2007). A precise balance of SCV-associated SipA levels appears to be critical for the stabilization of SCVs and their perinuclear positioning (Brawn et al. 2007). In this context, SipA is required for the proper localization on the SCVs of the Salmonella type SPI-2 effector SifA, another WXXXE effector described to antagonize the function of the Rab9 small GTPase and involved in the membrane tubulation of SCVs (Brawn et al. 2007; Jackson et al. 2008).
The Salmonella sip and Shigella ipa operons encode the T3SS translocator components and the SipA and IpaA orthologs. While SipA and IpaA share significant homology in the amino-terminal two-thirds of their primary sequence, the carboxyterminal region of SipA, corresponding to the F-actin-binding domain, differs from that of IpaA. Instead, the IpaA carboxyterminal region contains three binding sites for vinculin (Park et al. 2011). Vinculin is a cytoskeletal linker, which, upon activation, bridges integrin and cadherin receptors to the cytoskeleton (Atherton et al. 2016). Vinculin is composed of a globular head domain and a carboxyterminal tail domain containing an F-actin-binding site. Under its inactive form, intramolecular interactions between the vinculin head and tail domains maintain vinculin in a folded conformation where most ligand-binding sites are masked (Bakolitsa et al. 2004). Endogenous activators of vinculin, such as the focal adhesion protein talin, often contain several vinculin-binding sites (VBSs). For all VBSs described to date, vinculin activation occurs through binding to the amino-terminal region of the vinculin head domain, promoting major conformational changes that disrupt the head-tail intramolecular interactions, freeing the vinculin F-actin-binding region and enabling its binding to actin filaments (Izard et al. 2004). The IpaA carboxyterminals VBS1 and VBS2 act together as a super-mimic of endogenous VBSs. By binding to vinculin’s amino-terminal domain with an extremely high affinity, they promote vinculin activation (Nhieu and Izard 2007). The more proximal IpaA VBS3 functionally cooperates with IpaA VBS1 and VBS2 to trigger the recruitment of vinculin at bacterial invasion sites (Park et al. 2011). Through its VBSs, IpaA triggers the formation of an adhesion structure, stabilizing the bacteria at sites of invasion and potentially bypassing receptor-mediated anchorage (Valencia-Gallardo et al. 2015).
Recently, a domain in the Chlamydia type III effector TarP homologous to the vinculin-binding domain of IpaA was reported (Thwaites et al. 2015). As for IpaA, the Chlamydia vinculin-binding carboxyterminal domain contains three VBSs, with a similar organization and that appear to play similar roles as IpaA VBSs in triggering vinculin recruitment and bacterial invasion (Thwaites et al. 2015). TarP was also shown to interact in vitro with G- and F-actin via its carboxyterminal domain. Two carboxyterminal F-actin-binding TarP domains termed FAB1 and FAB2 were shown to mediate the bundling of actin filaments, an activity which may account for the formation of actin bundles underneath bound bacteria during the Chlamydia invasion process (Jiwani et al. 2013). TarP was initially reported as an actin nucleator, with the actin nucleation being mediated by a ≈200-residue proline-rich domain containing the G-actin-binding site with some similarity to the actin-binding site of WH2-family proteins (Jewett et al. 2006). Interestingly, the TarP proline-rich domain is also involved in TarP oligomerization, the latter being inseparable from the actin-nucleating activity. In vitro, TarP induces the formation of long unbranched filaments, through a mechanism that probably differs from those reported for the Arp2/3 complex or the endogenous actin nucleators formins and Spire (Jewett et al. 2006). The TarP proline-rich domain is essential for actin polymerization, but the precise role of its nucleating activity during Chlamydia invasion is not entirely clear, because, as discussed above, TarP-mediated actin polymerization requires Rac and Arp2/3 complex activation (Jewett et al. 2010). As for the Salmonella SipC translocon component, it has been postulated that following injection by the T3SS into host cells, the TarP actin-nucleating activity may serve to build an initial actin scaffold onto which Arp2/3-dependent actin nucleation would further expand. Interestingly, the number of tyrosine-rich repeats varies between C. trachomatis isolates, and these repeats are even absent from many Chlamydia species. The number of actin-binding domains also varies between species. These observations suggested that TarP is among the few known genes to play a role in C. trachomatis adaptations to specific niches within the host (Lutter et al. 2010). TarP was the first T3 effector of infectious Chlamydia to be identified and remains by far the best characterized one. Several other effectors are translocated together with TarP, and it is likely that they also contribute to the actin and membrane remodeling needed for invasion. For instance the effector CT694 interacts with the cytoskeletal organizing protein AHNAK, but how this contributes to bacterial uptake is not known (Hower et al. 2009).
8 Type III Effectors Interfering with Tyrosine Kinase Signaling
Pathogenic bacteria inject effectors into host cells that act as toxins, interfere with actin polymerization or actin filament organization. Such activities may serve to prevent phagocytosis by immune cells, downregulation of inflammatory signals, and adaptive Th1 immunity or to alter the endothelial or epithelial junctional integrity (Sarantis and Grinstein 2012). As for effectors involved in actin polymerization, these inhibitory effectors can act at the level of Rho GTPases or directly at the level of actin. Both these processes have been described in several excellent reviews (Aktories et al. 2011; Lemichez and Aktories 2013; Aktories et al, this issue). Tyrosine kinase signaling required for cytoskeletal reorganization is also targeted by type III effectors. Yersinia enterocolitica and Y. pseudotuberculosis are invasive enteropathogens that multiply extracellularly, following the crossing of the intestinal epithelial layer. Yersinia injects various type III effectors to prevent cytoskeletal reorganization required for phagocytosis at different levels of the signaling cascade. Among them, YopH is a highly efficient tyrosine phosphatase that appears to specifically target signaling downstream of β1-integrins, such as FAK, the Fak homolog Pyk, Cas, and paxillin (Viboud and Bliska 2005). In addition to the canonical tyrosine phosphatase motif C(X)5R(S/T) present on the P-loop, the substrate specificity of YopH is determined by two additional domains allowing its localization to cell adhesion structures and binding to tyrosyl-phosphorylated proteins (Ivanov et al. 2005). YopH-mediated inhibition of tyrosine kinase signaling thus prevents the activation of Rac and actin polymerization downstream of β1-integrin (Viboud and Bliska 2005). The targeting of β1-integrins by Yersinia via the invasin surface protein suggests a sophisticated pathogen-host cell interplay, whereby Yersinia hijacks β1-integrins to invade M cells and to prevent its phagocytosis by macrophages through the additional action of Yop proteins.
As discussed earlier, the Salmonella SptP also possesses a tyrosine phosphatase activity in addition to its RhoGAP function (Stebbins and Galan 2001). The substrate specificity of SptP is not clear at present. Structural characterization of SptP indicates that the tyrosine phosphatase domain is not affected by conformational changes in the carboxyterminal GAP domain (Stebbins and Galan 2001). This had led to speculations that, following targeting of Cdc42 or Rac via its GAP domain, SptP may specifically dephosphorylate substrates in complex with these GTPases.
EPEC also inhibits opsonized phagocytosis by macrophages via FcRgRIIa receptors through the action of the type III effector EspJ (Young et al. 2014). EspJ harbors a unique dual amidase-ADP-ribosyltransferase activity that modifies key residues in the catalytic domain of Src family kinases, required for FcRgRIIa phosphorylation and actin polymerization (Young et al. 2014). While the precise mechanism underlying the dual modification or residues remains undefined, mass spectroscopy analysis argues in favor of coupled, rather than sequential, amidation and ADP-ribosylation mediated by EspJ (Young et al. 2014). As for EspH, in addition to inhibiting bacterial phagocytosis by macrophages, EspJ may also be involved in the downregulation of pedestal formation during late phases of EPEC/EHEC infection (Wong et al. 2012; Young et al. 2014).
9 Concluding Remarks
In this chapter, we have tried to illustrate the wide diversity of strategies employed by bacterial pathogens to move intracellularly or to divert cytoskeletal processes using injected type III effectors. While being different, the comparison of the various strategies point to the targeting of converging hubs corresponding to tyrosine kinase signaling, RhoGTPases, phosphoinositides, and cytoskeletal proteins. Further investigation on other injected effectors reorganizing the actin cytoskeleton, as well as on their mechanisms of action, should allow to further deconvolve specific and fundamental features of each system. Because of the functional versatility of the actin cytoskeleton, much is probably yet to be learned about the manipulation of cytoskeletal processes by bacterial pathogens, beyond the initial epithelial/endothelial interactions. For example, relevant but less studied aspects include processes linked to the dissemination or egress of bacteria from infected tissue. Also, within mucosal surfaces and in addition to epithelial cells, pathogens also interact with immune cells, such as professional phagocytes, dendritic cells, macrophages, or neutrophils. Various pathogens have been reported to impair cytoskeletal responses involved in phagocytosis or lymphocyte activation. Because of their role in bacterial clearance or mounting of the inflammation, these interactions dictate the outcome of infection. Clearly, throughout the course of the infectious process, disease progression will critically depend on the pathogen’s ability to counter innate immunity.
References
Akin O, Mullins RD (2008) Capping protein increases the rate of actin-based motility by promoting filament nucleation by the Arp2/3 complex. Cell 133:841–851
Akopyan K, Edgren T, Wang-Edgren H, Rosqvist R, Fahlgren A, Wolf-Watz H, Fallman M (2011) Translocation of surface-localized effectors in type III secretion. Proc Natl Acad Sci U S A 108:1639–1644
Aktories K, Lang AE, Schwan C, Mannherz HG (2011) Actin as target for modification by bacterial protein toxins. FEBS J 278:4526–4543
Alto NM, Shao F, Lazar CS, Brost RL, Chua G, Mattoo S, McMahon SA, Ghosh P, Hughes TR, Boone C et al (2006) Identification of a bacterial type III effector family with G protein mimicry functions. Cell 124:133–145
Alto NM, Weflen AW, Rardin MJ, Yarar D, Lazar CS, Tonikian R, Koller A, Taylor SS, Boone C, Sidhu SS et al (2007) The type III effector EspF coordinates membrane trafficking by the spatiotemporal activation of two eukaryotic signaling pathways. J Cell Biol 178:1265–1278
Arbeloa A, Garnett J, Lillington J, Bulgin RR, Berger CN, Lea SM, Matthews S, Frankel G (2010) EspM2 is a RhoA guanine nucleotide exchange factor. Cell Microbiol 12:654–664
Atherton P, Stutchbury B, Jethwa D, Ballestrem C (2016) Mechanosensitive components of integrin adhesions: Role of vinculin. Exp Cell Res 343:21–27
Auerbuch V, Loureiro JJ, Gertler FB, Theriot JA, Portnoy DA (2003) Ena/VASP proteins contribute to Listeria monocytogenes pathogenesis by controlling temporal and spatial persistence of bacterial actin-based motility. Mol Microbiol 49:1361–1375
Bakolitsa C, Cohen DM, Bankston LA, Bobkov AA, Cadwell GW, Jennings L, Critchley DR, Craig SW, Liddington RC (2004) Structural basis for vinculin activation at sites of cell adhesion. Nature 430:583–586
Balana ME, Niedergang F, Subtil A, Alcover A, Chavrier P, Dautry-Varsat A (2005) ARF6 GTPase controls bacterial invasion by actin remodelling. J Cell Sci 118:2201–2210
Bastidas RJ, Elwell CA, Engel JN, Valdivia RH (2013) Chlamydial intracellular survival strategies. Cold Spring Harb Perspect Med 3:a010256
Bernardini ML, Mounier J, d’Hauteville H, Coquis-Rondon M, Sansonetti PJ (1989) Identification of icsA, a plasmid locus of Shigella flexneri that governs bacterial intra- and intercellular spread through interaction with F-actin. Proc Natl Acad Sci U S A 86:3867–3871
Bishai EA, Sidhu GS, Li W, Dhillon J, Bohil AB, Cheney RE, Hartwig JH, Southwick FS (2013) Myosin-X facilitates Shigella-induced membrane protrusions and cell-to-cell spread. Cell Microbiol 15:353–367
Bommarius B, Maxwell D, Swimm A, Leung S, Corbett A, Bornmann W, Kalman D (2007) Enteropathogenic Escherichia coli Tir is an SH2/3 ligand that recruits and activates tyrosine kinases required for pedestal formation. Mol Microbiol 63:1748–1768
Bornschlogl T, Romero S, Vestergaard CL, Joanny JF, Van Nhieu GT, Bassereau P (2013) Filopodial retraction force is generated by cortical actin dynamics and controlled by reversible tethering at the tip. Proc Natl Acad Sci U S A 110:18928–18933
Bougneres L, Girardin SE, Weed SA, Karginov AV, Olivo-Marin JC, Parsons JT, Sansonetti PJ, Van Nhieu GT (2004) Cortactin and Crk cooperate to trigger actin polymerization during Shigella invasion of epithelial cells. J Cell Biol 166:225–235
Boukellal H, Campas O, Joanny JF, Prost J, Sykes C (2004) Soft Listeria: actin-based propulsion of liquid drops. Phys Rev E Stat Nonlin Soft Matter Phys 69:061906
Brawn LC, Hayward RD, Koronakis V (2007) Salmonella SPI1 effector SipA persists after entry and cooperates with a SPI2 effector to regulate phagosome maturation and intracellular replication. Cell Host Microbe 1:63–75
Breitsprecher D, Kiesewetter AK, Linkner J, Vinzenz M, Stradal TE, Small JV, Curth U, Dickinson RB, Faix J (2011) Molecular mechanism of Ena/VASP-mediated actin-filament elongation. EMBO J 30:456–467
Bulgin R, Arbeloa A, Goulding D, Dougan G, Crepin VF, Raymond B, Frankel G (2009) The T3SS effector EspT defines a new category of invasive enteropathogenic E. coli (EPEC) which form intracellular actin pedestals. PLoS Pathog 5:e1000683
Burton EA, Plattner R, Pendergast AM (2003) Abl tyrosine kinases are required for infection by Shigella flexneri. EMBO J 22:5471–5479
Burton EA, Oliver TN, Pendergast AM (2005) Abl kinases regulate actin comet tail elongation via an N-WASP-dependent pathway. Mol Cell Biol 25:8834–8843
Campellone KG, Leong JM (2003) Tails of two Tirs: actin pedestal formation by enteropathogenic E. coli and enterohemorrhagic E. coli O157:H7. Curr Opin Microbiol 6:82–90
Clifton DR, Dooley CA, Grieshaber SS, Carabeo RA, Fields KA, Hackstadt T (2005) Tyrosine phosphorylation of the chlamydial effector protein Tarp is species specific and not required for recruitment of actin. Infect Immun 73:3860–3868
Cui J, Shao F (2011) Biochemistry and cell signaling taught by bacterial effectors. Trends Biochem Sci 36:532–540
Diakonova M, Helfer E, Seveau S, Swanson JA, Kocks C, Rui L, Carlier MF, Carter-Su C (2007) Adapter protein SH2-Bbeta stimulates actin-based motility of Listeria monocytogenes in a vasodilator-stimulated phosphoprotein (VASP)-dependent fashion. Infect Immun 75:3581–3593
Dragoi AM, Talman AM, Agaisse H (2013) Bruton’s tyrosine kinase regulates Shigella flexneri dissemination in HT-29 intestinal cells. Infect Immun 81:598–607
Egile C, Loisel TP, Laurent V, Li R, Pantaloni D, Sansonetti PJ, Carlier MF (1999) Activation of the CDC42 effector N-WASP by the Shigella flexneri IcsA protein promotes actin nucleation by Arp2/3 complex and bacterial actin-based motility. J Cell Biol 146:1319–1332
Elwell CA, Ceesay A, Kim JH, Kalman D, Engel JN (2008) RNA interference screen identifies Abl kinase and PDGFR signaling in Chlamydia trachomatis entry. PLoS Pathog 4, e1000021
Fallman M, Deleuil F, McGee K (2002) Resistance to phagocytosis by Yersinia. Int J Med Microbiol 291:501–509
Fattouh R, Kwon H, Czuczman MA, Copeland JW, Pelletier L, Quinlan ME, Muise AM, Higgins DE, Brumell JH (2015) The diaphanous-related formins promote protrusion formation and cell-to-cell spread of Listeria monocytogenes. J Infect Dis 211:1185–1195
Friebel A, Ilchmann H, Aepfelbacher M, Ehrbar K, Machleidt W, Hardt WD (2001) SopE and SopE2 from Salmonella typhimurium activate different sets of RhoGTPases of the host cell. J Biol Chem 276:34035–34040
Fukumatsu M, Ogawa M, Arakawa S, Suzuki M, Nakayama K, Shimizu S, Kim M, Mimuro H, Sasakawa C (2012) Shigella targets epithelial tricellular junctions and uses a noncanonical clathrin-dependent endocytic pathway to spread between cells. Cell Host Microbe 11:325–336
Galan JE, Lara-Tejero M, Marlovits TC, Wagner S (2014) Bacterial type III secretion systems: specialized nanomachines for protein delivery into target cells. Annu Rev Microbiol 68:415–438
Garber JJ, Takeshima F, Anton IM, Oyoshi MK, Lyubimova A, Kapoor A, Shibata T, Chen F, Alt FW, Geha RS et al (2012) Enteropathogenic Escherichia coli and vaccinia virus do not require the family of WASP-interacting proteins for pathogen-induced actin assembly. Infect Immun 80:4071–4077
Garza-Mayers AC, Miller KA, Russo BC, Nagda DV, Goldberg MB (2015) Shigella flexneri regulation of ARF6 activation during bacterial entry via an IpgD-mediated positive feedback loop. MBio 6, e02584
Gerbal F, Laurent V, Ott A, Carlier MF, Chaikin P, Prost J (2000) Measurement of the elasticity of the actin tail of Listeria monocytogenes. Eur Biophys J 29:134–140
Gouin E, Gantelet H, Egile C, Lasa I, Ohayon H, Villiers V, Gounon P, Sansonetti PJ, Cossart P (1999) A comparative study of the actin-based motilities of the pathogenic bacteria Listeria monocytogenes, Shigella flexneri and Rickettsia conorii. J Cell Sci 112(Pt 11):1697–1708
Hachani A, Biskri L, Rossi G, Marty A, Menard R, Sansonetti P, Parsot C, Van Nhieu GT, Bernardini ML, Allaoui A (2008) IpgB1 and IpgB2, two homologous effectors secreted via the Mxi-Spa type III secretion apparatus, cooperate to mediate polarized cell invasion and inflammatory potential of Shigella flexenri. Microbes Infect 10:260–268
Haglund CM, Choe JE, Skau CT, Kovar DR, Welch MD (2010) Rickettsia Sca2 is a bacterial formin-like mediator of actin-based motility. Nat Cell Biol 12:1057–1063
Handa Y, Suzuki M, Ohya K, Iwai H, Ishijima N, Koleske AJ, Fukui Y, Sasakawa C (2007) Shigella IpgB1 promotes bacterial entry through the ELMO-Dock180 machinery. Nat Cell Biol 9:121–128
Hanisch J, Kolm R, Wozniczka M, Bumann D, Rottner K, Stradal TE (2011) Activation of a RhoA/myosin II-dependent but Arp2/3 complex-independent pathway facilitates Salmonella invasion. Cell Host Microbe 9:273–285
Hardt WD, Chen LM, Schuebel KE, Bustelo XR, Galan JE (1998) S. typhimurium encodes an activator of Rho GTPases that induces membrane ruffling and nuclear responses in host cells. Cell 93:815–826
Harlander RS, Way M, Ren Q, Howe D, Grieshaber SS, Heinzen RA (2003) Effects of ectopically expressed neuronal Wiskott-Aldrich syndrome protein domains on Rickettsia rickettsii actin-based motility. Infect Immun 71:1551–1556
Hayward RD, Koronakis V (1999) Direct nucleation and bundling of actin by the SipC protein of invasive Salmonella. EMBO J 18:4926–4934
Heindl JE, Saran I, Yi CR, Lesser CF, Goldberg MB (2010) Requirement for formin-induced actin polymerization during spread of Shigella flexneri. Infect Immun 78:193–203
Ho HY, Rohatgi R, Lebensohn AM, Le M, Li J, Gygi SP, Kirschner MW (2004) Toca-1 mediates Cdc42-dependent actin nucleation by activating the N-WASP-WIP complex. Cell 118:203–216
Hoffmann AK, Naj X, Linder S (2014) Daam1 is a regulator of filopodia formation and phagocytic uptake of Borrelia burgdorferi by primary human macrophages. FASEB J 28:3075–3089
Hower S, Wolf K, Fields KA (2009) Evidence that CT694 is a novel Chlamydia trachomatis T3S substrate capable of functioning during invasion or early cycle development. Mol Microbiol 72:1423–1437
Huang Z, Sutton SE, Wallenfang AJ, Orchard RC, Wu X, Feng Y, Chai J, Alto NM (2009) Structural insights into host GTPase isoform selection by a family of bacterial GEF mimics. Nat Struct Mol Biol 16:853–860
Humphreys D, Davidson A, Hume PJ, Koronakis V (2012) Salmonella virulence effector SopE and Host GEF ARNO cooperate to recruit and activate WAVE to trigger bacterial invasion. Cell Host Microbe 11:129–139
Ivanov MI, Stuckey JA, Schubert HL, Saper MA, Bliska JB (2005) Two substrate-targeting sites in the Yersinia protein tyrosine phosphatase co-operate to promote bacterial virulence. Mol Microbiol 55:1346–1356
Izard T, Evans G, Borgon RA, Rush CL, Bricogne G, Bois PR (2004) Vinculin activation by talin through helical bundle conversion. Nature 427:171–175
Jackson LK, Nawabi P, Hentea C, Roark EA, Haldar K (2008) The Salmonella virulence protein SifA is a G protein antagonist. Proc Natl Acad Sci U S A 105:14141–14146
Jasnin M, Asano S, Gouin E, Hegerl R, Plitzko JM, Villa E, Cossart P, Baumeister W (2013) Three-dimensional architecture of actin filaments in Listeria monocytogenes comet tails. Proc Natl Acad Sci U S A 110:20521–20526
Jeng RL, Goley ED, D’Alessio JA, Chaga OY, Svitkina TM, Borisy GG, Heinzen RA, Welch MD (2004) A Rickettsia WASP-like protein activates the Arp2/3 complex and mediates actin-based motility. Cell Microbiol 6:761–769
Jepson MA, Pellegrin S, Peto L, Banbury DN, Leard AD, Mellor H, Kenny B (2003) Synergistic roles for the Map and Tir effector molecules in mediating uptake of enteropathogenic Escherichia coli (EPEC) into non-phagocytic cells. Cell Microbiol 5:773–783
Jewett TJ, Fischer ER, Mead DJ, Hackstadt T (2006) Chlamydial TARP is a bacterial nucleator of actin. Proc Natl Acad Sci U S A 103:15599–15604
Jewett TJ, Dooley CA, Mead DJ, Hackstadt T (2008) Chlamydia trachomatis tarp is phosphorylated by src family tyrosine kinases. Biochem Biophys Res Commun 371:339–344
Jewett TJ, Miller NJ, Dooley CA, Hackstadt T (2010) The conserved Tarp actin binding domain is important for chlamydial invasion. PLoS Pathog 6, e1000997
Jiwani S, Alvarado S, Ohr RJ, Romero A, Nguyen B, Jewett TJ (2013) Chlamydia trachomatis Tarp harbors distinct G and F actin binding domains that bundle actin filaments. J Bacteriol 195:708–716
Kenny B, Ellis S, Leard AD, Warawa J, Mellor H, Jepson MA (2002) Co-ordinate regulation of distinct host cell signalling pathways by multifunctional enteropathogenic Escherichia coli effector molecules. Mol Microbiol 44:1095–1107
Kocks C, Gouin E, Tabouret M, Berche P, Ohayon H, Cossart P (1992) L. monocytogenes-induced actin assembly requires the actA gene product, a surface protein. Cell 68:521–531
Konradt C, Frigimelica E, Nothelfer K, Puhar A, Salgado-Pabon W, di Bartolo V, Scott-Algara D, Rodrigues CD, Sansonetti PJ, Phalipon A (2011) The Shigella flexneri type three secretion system effector IpgD inhibits T cell migration by manipulating host phosphoinositide metabolism. Cell Host Microbe 9:263–272
Krause M, Gautreau A (2014) Steering cell migration: lamellipodium dynamics and the regulation of directional persistence. Nat Rev Mol Cell Biol 15:577–590
Kubori T, Galan JE (2003) Temporal regulation of salmonella virulence effector function by proteasome-dependent protein degradation. Cell 115:333–342
Kuehl CJ, Dragoi AM, Talman A, Agaisse H (2015) Bacterial spread from cell to cell: beyond actin-based motility. Trends Microbiol 23:558–566
Lai Y, Rosenshine I, Leong JM, Frankel G (2013) Intimate host attachment: enteropathogenic and enterohaemorrhagic Escherichia coli. Cell Microbiol 15:1796–1808
Lane BJ, Mutchler C, Al Khodor S, Grieshaber SS, Carabeo RA (2008) Chlamydial entry involves TARP binding of guanine nucleotide exchange factors. PLoS Pathog 4, e1000014
Lehmann MJ, Sherer NM, Marks CB, Pypaert M, Mothes W (2005) Actin- and myosin-driven movement of viruses along filopodia precedes their entry into cells. J Cell Biol 170:317–325
Lemichez E, Aktories K (2013) Hijacking of Rho GTPases during bacterial infection. Exp Cell Res 319:2329–2336
Leung Y, Ally S, Goldberg MB (2008) Bacterial actin assembly requires toca-1 to relieve N-wasp autoinhibition. Cell Host Microbe 3:39–47
Lilic M, Galkin VE, Orlova A, VanLoock MS, Egelman EH, Stebbins CE (2003) Salmonella SipA polymerizes actin by stapling filaments with nonglobular protein arms. Science 301:1918–1921
Loisel TP, Boujemaa R, Pantaloni D, Carlier MF (1999) Reconstitution of actin-based motility of Listeria and Shigella using pure proteins. Nature 401:613–616
Lum M, Morona R (2014) Myosin IIA is essential for Shigella flexneri cell-to-cell spread. Pathog Dis 72:174–187
Lutter EI, Bonner C, Holland MJ, Suchland RJ, Stamm WE, Jewett TJ, McClarty G, Hackstadt T (2010) Phylogenetic analysis of Chlamydia trachomatis Tarp and correlation with clinical phenotype. Infect Immun 78:3678–3688
Ly KT, Casanova JE (2009) Abelson tyrosine kinase facilitates Salmonella enterica serovar Typhimurium entry into epithelial cells. Infect Immun 77:60–69
Mallo GV, Espina M, Smith AC, Terebiznik MR, Aleman A, Finlay BB, Rameh LE, Grinstein S, Brumell JH (2008) SopB promotes phosphatidylinositol 3-phosphate formation on Salmonella vacuoles by recruiting Rab5 and Vps34. J Cell Biol 182:741–752
McGhie EJ, Hayward RD, Koronakis V (2001) Cooperation between actin-binding proteins of invasive Salmonella: SipA potentiates SipC nucleation and bundling of actin. EMBO J 20:2131–2139
Mehlitz A, Rudel T (2013) Modulation of host signaling and cellular responses by Chlamydia. Cell Commun Signal 11:90
Mital J, Hackstadt T (2011) Diverse requirements for SRC-family tyrosine kinases distinguish chlamydial species. MBio 2
Mogilner A, Oster G (1996) Cell motility driven by actin polymerization. Biophys J 71:3030–3045
Mogilner A, Oster G (2003) Force generation by actin polymerization II: the elastic ratchet and tethered filaments. Biophys J 84:1591–1605
Nhieu GT, Izard T (2007) Vinculin binding in its closed conformation by a helix addition mechanism. EMBO J 26:4588–4596
Niebuhr K, Ebel F, Frank R, Reinhard M, Domann E, Carl UD, Walter U, Gertler FB, Wehland J, Chakraborty T (1997) A novel proline-rich motif present in ActA of Listeria monocytogenes and cytoskeletal proteins is the ligand for the EVH1 domain, a protein module present in the Ena/VASP family. EMBO J 16:5433–5444
Niebuhr K, Giuriato S, Pedron T, Philpott DJ, Gaits F, Sable J, Sheetz MP, Parsot C, Sansonetti PJ, Payrastre B (2002) Conversion of PtdIns(4,5)P(2) into PtdIns(5)P by the S. flexneri effector IpgD reorganizes host cell morphology. EMBO J 21:5069–5078
Orchard RC, Alto NM (2012) Mimicking GEFs: a common theme for bacterial pathogens. Cell Microbiol 14:10–18
Pantaloni D, Le Clainche C, Carlier MF (2001) Mechanism of actin-based motility. Science 292:1502–1506
Park H, Valencia-Gallardo C, Sharff A, Tran Van Nhieu G, Izard T (2011) Novel vinculin binding site of the IpaA invasin of Shigella. J Biol Chem 286:23214–23221
Patel JC, Galan JE (2006) Differential activation and function of Rho GTPases during Salmonella-host cell interactions. J Cell Biol 175:453–463
Picking WL, Picking WD (2016) The many faces of IpaB. Front Cell Infect Microbiol 6:12
Piscatelli HL, Li M, Zhou D (2016) Dual 4- and 5-phosphatase activities regulate SopB-dependent phosphoinositide dynamics to promote bacterial entry. Cell Microbiol 18:705–719
Rajabian T, Gavicherla B, Heisig M, Muller-Altrock S, Goebel W, Gray-Owen SD, Ireton K (2009) The bacterial virulence factor InlC perturbs apical cell junctions and promotes cell-to-cell spread of Listeria. Nat Cell Biol 11:1212–1218
Rathman M, de Lanerolle P, Ohayon H, Gounon P, Sansonetti P (2000) Myosin light chain kinase plays an essential role in S. flexneri dissemination. J Cell Sci 113 Pt 19:3375–3386
Reed SC, Lamason RL, Risca VI, Abernathy E, Welch MD (2014) Rickettsia actin-based motility occurs in distinct phases mediated by different actin nucleators. Curr Biol 24:98–103
Reinhard M, Giehl K, Abel K, Haffner C, Jarchau T, Hoppe V, Jockusch BM, Walter U (1995) The proline-rich focal adhesion and microfilament protein VASP is a ligand for profilins. EMBO J 14:1583–1589
Rohatgi R, Ma L, Miki H, Lopez M, Kirchhausen T, Takenawa T, Kirschner MW (1999) The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell 97:221–231
Romero S, Grompone G, Carayol N, Mounier J, Guadagnini S, Prevost MC, Sansonetti PJ, Van Nhieu GT (2011) ATP-mediated Erk1/2 activation stimulates bacterial capture by filopodia, which precedes Shigella invasion of epithelial cells. Cell Host Microbe 9:508–519
Samarin S, Romero S, Kocks C, Didry D, Pantaloni D, Carlier MF (2003) How VASP enhances actin-based motility. J Cell Biol 163:131–142
Santy LC, Ravichandran KS, Casanova JE (2005) The DOCK180/Elmo complex couples ARNO-mediated Arf6 activation to the downstream activation of Rac1. Curr Biol 15:1749–1754
Sarantis H, Grinstein S (2012) Subversion of phagocytosis for pathogen survival. Cell Host Microbe 12:419–431
Sason H, Milgrom M, Weiss AM, Melamed-Book N, Balla T, Grinstein S, Backert S, Rosenshine I, Aroeti B (2009) Enteropathogenic Escherichia coli subverts phosphatidylinositol 4,5-bisphosphate and phosphatidylinositol 3,4,5-trisphosphate upon epithelial cell infection. Mol Biol Cell 20:544–555
Shaw RK, Cleary J, Murphy MS, Frankel G, Knutton S (2005) Interaction of enteropathogenic Escherichia coli with human intestinal mucosa: role of effector proteins in brush border remodeling and formation of attaching and effacing lesions. Infect Immun 73:1243–1251
Shi J, Casanova JE (2006) Invasion of host cells by Salmonella typhimurium requires focal adhesion kinase and p130Cas. Mol Biol Cell 17:4698–4708
Simovitch M, Sason H, Cohen S, Zahavi EE, Melamed-Book N, Weiss A, Aroeti B, Rosenshine I (2010) EspM inhibits pedestal formation by enterohaemorrhagic Escherichia coli and enteropathogenic E. coli and disrupts the architecture of a polarized epithelial monolayer. Cell Microbiol 12:489–505
Smith K, Humphreys D, Hume PJ, Koronakis V (2010) Enteropathogenic Escherichia coli recruits the cellular inositol phosphatase SHIP2 to regulate actin-pedestal formation. Cell Host Microbe 7:13–24
Soo FS, Theriot JA (2005) Large-scale quantitative analysis of sources of variation in the actin polymerization-based movement of Listeria monocytogenes. Biophys J 89:703–723
Stebbins CE, Galan JE (2001) Structural mimicry in bacterial virulence. Nature 412:701–705
Stevens JM, Galyov EE, Stevens MP (2006) Actin-dependent movement of bacterial pathogens. Nat Rev Microbiol 4:91–101
Suei S, Seyan R, Noguera P, Manzi J, Plastino J, Kreplak L (2011) The mechanical role of VASP in an Arp2/3-complex-based motility assay. J Mol Biol 413:573–583
Suzuki T, Miki H, Takenawa T, Sasakawa C (1998) Neural Wiskott-Aldrich syndrome protein is implicated in the actin-based motility of Shigella flexneri. EMBO J 17:2767–2776
Suzuki T, Mimuro H, Suetsugu S, Miki H, Takenawa T, Sasakawa C (2002) Neural Wiskott-Aldrich syndrome protein (N-WASP) is the specific ligand for Shigella VirG among the WASP family and determines the host cell type allowing actin-based spreading. Cell Microbiol 4:223–233
Swimm A, Bommarius B, Reeves P, Sherman M, Kalman D (2004) Complex kinase requirements for EPEC pedestal formation. Nat Cell Biol 6:795, author reply 795-796
Theriot JA, Rosenblatt J, Portnoy DA, Goldschmidt-Clermont PJ, Mitchison TJ (1994) Involvement of profilin in the actin-based motility of L. monocytogenes in cells and in cell-free extracts. Cell 76:505–517
Thwaites T, Nogueira AT, Campeotto I, Silva AP, Grieshaber SS, Carabeo RA (2014) The Chlamydia effector TarP mimics the mammalian leucine-aspartic acid motif of paxillin to subvert the focal adhesion kinase during invasion. J Biol Chem 289:30426–30442
Thwaites TR, Pedrosa AT, Peacock TP, Carabeo RA (2015) Vinculin interacts with the chlamydia effector TarP via a tripartite vinculin binding domain to mediate actin recruitment and assembly at the plasma membrane. Front Cell Infect Microbiol 5:88
Truong D, Brabant D, Bashkurov M, Wan LC, Braun V, Heo WD, Meyer T, Pelletier L, Copeland J, Brumell JH (2013) Formin-mediated actin polymerization promotes Salmonella invasion. Cell Microbiol 15:2051–2063
Truong D, Copeland JW, Brumell JH (2014) Bacterial subversion of host cytoskeletal machinery: hijacking formins and the Arp2/3 complex. Bioessays 36:687–696
Valdez Y, Ferreira RB, Finlay BB (2009) Molecular mechanisms of Salmonella virulence and host resistance. Curr Top Microbiol Immunol 337:93–127
Valencia-Gallardo CM, Carayol N, Tran Van Nhieu G (2015) Cytoskeletal mechanics during Shigella invasion and dissemination in epithelial cells. Cell Microbiol 17:174–182
Van Engelenburg SB, Palmer AE (2008) Quantification of real-time Salmonella effector type III secretion kinetics reveals differential secretion rates for SopE2 and SptP. Chem Biol 15:619–628
Van Kirk LS, Hayes SF, Heinzen RA (2000) Ultrastructure of Rickettsia rickettsii actin tails and localization of cytoskeletal proteins. Infect Immun 68:4706–4713
Viaud J, Lagarrigue F, Ramel D, Allart S, Chicanne G, Ceccato L, Courilleau D, Xuereb JM, Pertz O, Payrastre B et al (2014) Phosphatidylinositol 5-phosphate regulates invasion through binding and activation of Tiam1. Nat Commun 5:4080
Viaud J, Mansour R, Antkowiak A, Mujalli A, Valet C, Chicanne G, Xuereb JM, Terrisse AD, Severin S, Gratacap MP et al (2016) Phosphoinositides: important lipids in the coordination of cell dynamics. Biochimie 125:250–258
Viboud GI, Bliska JB (2005) Yersinia outer proteins: role in modulation of host cell signaling responses and pathogenesis. Annu Rev Microbiol 59:69–89
Wang J, King JE, Goldrick M, Lowe M, Gertler FB, Roberts IS (2015) Lamellipodin is important for cell-to-cell spread and actin-based motility in Listeria monocytogenes. Infect Immun 83:3740–3748
Welch MD, Way M (2013) Arp2/3-mediated actin-based motility: a tail of pathogen abuse. Cell Host Microbe 14:242–255
Welch MD, Iwamatsu A, Mitchison TJ (1997) Actin polymerization is induced by Arp2/3 protein complex at the surface of Listeria monocytogenes. Nature 385:265–269
Welch MD, Rosenblatt J, Skoble J, Portnoy DA, Mitchison TJ (1998) Interaction of human Arp2/3 complex and the Listeria monocytogenes ActA protein in actin filament nucleation. Science 281:105–108
Wessler S, Backert S (2011) Abl family of tyrosine kinases and microbial pathogenesis. Int Rev Cell Mol Biol 286:271–300
Westermark L, Fahlgren A, Fallman M (2014) Yersinia pseudotuberculosis efficiently escapes polymorphonuclear neutrophils during early infection. Infect Immun 82:1181–1191
Wiesner S, Helfer E, Didry D, Ducouret G, Lafuma F, Carlier MF, Pantaloni D (2003) A biomimetic motility assay provides insight into the mechanism of actin-based motility. J Cell Biol 160:387–398
Willcocks SJ, Denman CC, Atkins HS, Wren BW (2016) Intracellular replication of the well-armed pathogen Burkholderia pseudomallei. Curr Opin Microbiol 29:94–103
Wong AR, Pearson JS, Bright MD, Munera D, Robinson KS, Lee SF, Frankel G, Hartland EL (2011) Enteropathogenic and enterohaemorrhagic Escherichia coli: even more subversive elements. Mol Microbiol 80:1420–1438
Wong AR, Clements A, Raymond B, Crepin VF, Frankel G (2012) The interplay between the Escherichia coli Rho guanine nucleotide exchange factor effectors and the mammalian RhoGEF inhibitor EspH. MBio 3
Young VB, Falkow S, Schoolnik GK (1992) The invasin protein of Yersinia enterocolitica: internalization of invasin-bearing bacteria by eukaryotic cells is associated with reorganization of the cytoskeleton. J Cell Biol 116:197–207
Young JC, Clements A, Lang AE, Garnett JA, Munera D, Arbeloa A, Pearson J, Hartland EL, Matthews SJ, Mousnier A et al (2014) The Escherichia coli effector EspJ blocks Src kinase activity via amidation and ADP ribosylation. Nat Commun 5:5887
Zhou D, Chen LM, Hernandez L, Shears SB, Galan JE (2001) A Salmonella inositol polyphosphatase acts in conjunction with other bacterial effectors to promote host cell actin cytoskeleton rearrangements and bacterial internalization. Mol Microbiol 39:248–259
Acknowledgments
The authors thank Drs. Agathe Subtil and Olivera Francetic (Institut Pasteur, Paris, France) for proofreading the manuscript and Dr. John Leong (Tufts University, Boston, USA) for the helpful discussion. The work was supported by grants from Inserm, the CNRS, and the Collège de France to the CIRB, as well as a grant from the PSL Idex project “Shigaforce” (ANR-10-IDEX-0001-02 PSL*).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Van Nhieu, G.T., Romero, S. (2016). Common Themes in Cytoskeletal Remodeling by Intracellular Bacterial Effectors. In: Jockusch, B. (eds) The Actin Cytoskeleton. Handbook of Experimental Pharmacology, vol 235. Springer, Cham. https://doi.org/10.1007/164_2016_42
Download citation
DOI: https://doi.org/10.1007/164_2016_42
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-46369-8
Online ISBN: 978-3-319-46371-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)