
Abstract
This review gives representative examples of the various types of synthetic cationic polymers or polyampholytes (chemical structure, architecture, etc) that can be used to complex DNA (forming polyplexes) for their application in gene delivery. In designing polycations for gene delivery, one has to take into account a balance between protection of DNA versus loss of efficiency for DNA condensation and efficient condensation versus hindering of DNA release. Indeed, if the polyplexes are not stable enough, premature dissociation will occur before delivery of the genetic material at the desired place, resulting in low transfection efficiency; on the other hand, a complex that is too stable will not release the DNA, also resulting in low gene expression. The techniques generally used to determine these properties are gel electrophoresis to test the DNA/polymer complexation, ethidium bromide or polyanion displacement to test the affinity of a polymer for DNA, and light scattering to determine the extent of DNA condensation. Moreover, with the development of more precise instruments for physico-chemical characterization and appropriate biochemical and biophysical techniques, a direct link between the physico-chemical characteristics of the polyplexes and their in vitro and in vivo properties can be drawn, thus allowing tremendous progress in the quest towards application of polyplexes for gene therapy, beyond the research laboratory.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Colloidal stabilization
- DNA
- Gene delivery vectors
- Polyampholytes
- Polycations
- Polyelectrolyte complexes
- Polyplexes
1 Introduction
The subject of this review is complexes of DNA with synthetic cationic polymers and their application in gene delivery [1–4]. Linear, graft, and comb polymers (flexible, i.e., non-conjugated polymers) are its focus. This review is not meant to be exhaustive but to give representative examples of the various types (chemical structure, architecture, etc.) of synthetic cationic polymers or polyampholytes that can be used to complex DNA. Other interesting synthetic architectures such dendrimers [5–7], dendritic structures/polymers [8, 9], and hyperbranched polymers [10–12] will not be addressed because there are numerous recent valuable reports about their complexes with DNA. Natural or partially synthetic polymers such as polysaccharides (chitosan [13], dextran [14, 15], etc.) and peptides [16, 17] for DNA complexation or delivery will not be mentioned.
Since the first generation of polycations for cell transfection, such as poly(ethylene imine) (PEI, commercially available as ExGen500 or jetPEI in its linear form or as Lipofectamine, which is hyperbranched PEI incorporated in cationic lipids) [18, 19], poly(l-lysine) (PLL) [20], poly(amido amine) (PAMAM, Starburst) [8], poly(propylene imine) (PPI) [21, 22], and their derivatives, various other architectures and structural motifs have been designed in order to surpass the efficiency of these commercial products but unfortunately none of them have succeeded [23]. To date, no gene carrier has been approved for use in vivo despite the increasing numbers of clinical trials in this direction worldwide, and therefore research in the field of polycations as non-viral gene delivery vectors is still of prime importance.
It is to be noted that not only water-soluble polymers can be used to complex DNA, amphiphilic polymers, which depending on the relative ratio of hydrophilic to hydrophobic block, can also form various self-assembled structures, from spherical micelles to vesicles (polymersomes). This review will be restricted to micelle-forming polymers and will exclude polymersomes, which can both encapsulate (in their aqueous interior) and complex DNA [24, 25].
Since the early years of DNA complexation with cationic polymers, pioneers from the field of polyelectrolyte complexes between surfactants and/or polymers led solid physico-chemical studies on the complexation of DNA with polymers. But it is only more recently, with the rise of more precise instruments for physico-chemical characterization and appropriate biochemical and biophysical techniques, that the published studies are allowing a direct link to be drawn between physico-chemical characteristics (such as size, charge, etc.) of the DNA/polymer (mostly polycations) complexes, also called polyplexes, and their in vitro and in vivo properties, thus allowing tremendous progresses in the quest towards polyplexes for gene therapy and their application beyond research laboratories.
The Introduction will give a brief description of DNA as a biopolymer (structure, conformations, topologies), some definitions in the field of polyelectrolytes (weak and strong polyelectrolytes), some generalities about DNA/polycation complexes (factors influencing the complexation, models describing the structure of the polyplexes, methods adapted to their characterization), and a description of the parameters to take into consideration for their use in gene therapy.
Then in Sect. 2, the interpolyelectrolyte complexes (IPEC) between polycationic polymers and DNA will be addressed as a function of the chemical structure of the polymer (most of the DNA being used is plasmid DNA, consisting of many thousands of base pairs). Water-soluble and amphiphilic polymers will be discussed and then other properties will be taken into consideration such as the polyelectrolyte’s nature (strong or weak), the presence of steric stabilizers, etc. Section 3 will deal with complexes of polyamphoteric polymers with DNA. In both parts, the working line is the correlation between physico-chemical properties and efficiency in vitro (transfection potency).
Finally, we will give some perspectives on the field opened by new polymerization techniques, and consequently new types of polymers, and on recent discoveries about how to interfere with the expression of specific genes with oligonucleotides.
1.1 DNA
1.1.1 Structure of DNA
DNA (deoxyribonucleic acid) is a biopolymer containing the genetic information [26, 27]. Deoxyribonucleotides are the monomers of DNA and are all composed of three parts: a nitrogenous base also called nucleobase, a deoxyribose sugar, and one phosphate group (negatively charged at physiological pH). The nucleobase is always bound to the 1′-carbon of the deoxyribose and the phosphate groups bind to the 5′-carbon of the sugar. There are four different nucleobases: two purines [adenine (A) and guanine (G)], and two pyrimidines [cytosine (C) and thymine (T)]. The deoxyribonucleotides are linked with one another via 3′–5′-phosphodiester bounds.
DNA is composed of two antiparallel complementary strands, which build a double helix. Pairing of the bases, which grant stability to the helix, takes place via hydrogen bonds. The base pairs (bp) are A–T (two bonds) and G–C (three bonds), and constitute the inner side of the double helix (Scheme 1). The backbone of the helix is composed of the sugar-phosphate chain. Another important contribution to the stability of the helix comes from the base stacking of the aromatic rings of the nucleobases. The length of the strands also plays a role: the longer the strands (i.e., the more nucleobases there are to interact), the more stable is the double helix.

Chemical structure of DNA
1.1.2 Conformations of DNA
DNA can have different conformations (A-, B-, or Z-DNA), which vary in handedness, number of base pairs per helix turn, and diameter as proved by X-ray diffraction studies [28]. DNA in its native state is a semi-flexible long thin rod, only about 2 nm in diameter (B- and Z-DNA), with a persistence length (mechanical property quantifying the stiffness of a polymer) of about 50 nm [29, 30], which depends on ionic strength [31], DNA sequence [32], and temperature [33].
The conformation of the double helix can be studied using various spectroscopic methods such as circular dichroism (CD) [34], infrared (IR), Raman, ultraviolet (UV), visible absorption spectroscopy, and nuclear magnetic resonance (NMR) spectroscopy [35].
1.1.3 Topologies of DNA
DNA can be chromosomal or extra-chromosomal (plasmid DNA). Plasmid DNA (pDNA) is a double-stranded DNA (dsDNA) that can replicate independently of the chromosomal DNA, and is usually constituted of hundreds to a few thousand base pairs. Artificial plasmids are widely used in gene therapy in order to drive the replication of recombinant DNA sequences within host organisms. pDNA can adopt various conformations (linear, circular, or supercoiled) according to the over- or underwinding of a DNA strand (Scheme 2). DNA supercoiling is important for DNA packaging within all cells. Because the length of DNA can be thousands of times that of a cell, supercoiling of DNA allows DNA compaction, therefore much more genetic material can be packaged into the cell or nucleus (in eukaryotes).

Various topologies of dsDNA molecule: linear, circular, and supercoiled
The commercial calf thymus DNA (ctDNA) often used in physico-chemical studies is a linear DNA that can be isolated from calf thymus, an organ that has a very high yield of DNA.
The various topologies of DNA (supercoiled, circular, linear) can be discriminated by various methods such as electrophoresis and by microscopy techniques such as electron microscopy (EM) [36], cryogenic transmission electron microscopy (cryo-TEM) [37], and atomic force microscopy (AFM) [38].
1.1.4 DNA Condensation in Nature
Interpolyelectrolyte complexes form spontaneously upon mixing of solutions of oppositely charged polyelectrolytes, the main driving force being the gain of entropy because of the release of small counterions as well as the electrostatic interactions. This entropy-driven process creates an exceedingly tricky problem of how to package the genetic material in a stable non-aggregating form with synthetic polymers. On the other hand, the way Nature deals with the complexation of genetic material by proteins is extremely efficient: the genome of eukaryotic cells is packaged in a topologically controlled manner in the form of fibrous superstructures known as chromatin, and this allows DNA with a contour length of 2 m to be packaged in the nucleus of cells only a few micrometers in diameter [39]. The nucleosome core particle (NCP) is the fundamental building block of chromatin and contains approximatively 147 bp of DNA wrapped in roughly two superhelical turns around an octamer of four core histones (H2A, H2B, H3, H4) (Scheme 3b): the DNA that links two neighboring nucleosomes is called linker DNA (55 bp) [40]. The structure adopts a “beads-on-a-string” configuration (Scheme 3c).

Schematic view of some levels of DNA folding in the cell. (a) On length scales much smaller than the persistence length (p b ), DNA can be considered straight. (b) In eukaryotic cells, DNA wraps around a core of histone proteins to create a nucleosome structure. (c) Structure of chromation with “beads-on-a-string” configuration. Reprinted with permission from [243]. Copyright 2012 American Society for Biochemistry and Molecular Biology
The H1 protein interacts with NCPs and organizes linker DNA, helping stabilize the zig-zagged 30 nm chromatin fiber. This is a nice example found in Nature of controlled complexation of genetic material [negatively charged DNA and histones, constituted mainly of positively charged amino acids such as arginine (Arg) and lysine (Lys)].
The selective binding of a protein to a particular DNA sequence requires the recognition by the protein of an ensemble of steric and chemical features that delineate the binding site [41]. DNA–protein recognition occurs very often by insertion of an R-helix into the major groove of dsDNA. A specific DNA sequence is then recognized through:
-
1.
Formation of extensive hydrogen bonding and van der Waals interactions with the bases (“direct readout”)
-
2.
Recognition of sequence-dependent conformational features through electrostatic interactions with the negatively charged phosphodiester backbone (“indirect readout”)
The structure of these DNA-binding proteins and the way they bind to DNA can be taken as inspiration for the rational design of synthetic polymers as DNA complexants.
1.2 Polyelectrolytes
Due to the presence of negatively charged phosphate groups, DNA is a strong polyanion and can forms complexes with positively charged polymers. DNA is usually defined by its number of base pairs and molecular weight (in Daltons) per charge (two charges per bp, ~650 Da/bp). It is important to mention that the polyelectrolyte character of DNA largely controls its behavior in solution.
1.2.1 Weak and Strong Polyelectrolytes
Polyelectrolytes are polymers whose repeating units bear an ionizable group. These groups will dissociate in aqueous solutions, making the polymers charged. Polyelectrolytes can be divided into weak and strong polyelectrolytes. Strong polyelectrolytes dissociate completely in solution for most reasonable pH values, whereas weak polyelectrolytes have a dissociation constant (pK a) in the range of ~2 to 10, meaning that they will be partially dissociated at intermediate pH.
In the case of strong polyelectrolytes, the number and position of charges is fixed; variation of pH or ion concentration will not affect the number of charges. On the other hand, weak polyelectrolytes are not fully charged in solution, and their average degree of charges is given by the dissociation–association equilibrium constant and is governed by the pH of the solution, counterion concentration, and ionic strength; the charges are mobile within the polyelectrolyte.
The conformation of any polymer is affected by a number of factors, including the polymer architecture and the solvent affinity. In the case of polyelectrolytes, an additional factor is present: charge [42, 43]. In solution, whereas an uncharged linear polymer chain is usually found in a random conformation (theta solvent), a linear polyelectrolyte will adopt a more expanded, rigid-rod-like conformation due to the coulomb repulsion (the charges on the chain will repel each other) (Scheme 4a).

Counterion condensation on a polyelectrolyte
The structure of the polyelectrolyte itself depends on the grafting density, degree of dissociation with counterions, and ionic strength of the medium. If the ionic strength of a solution is high enough, the charges will be screened and consequently the polyelectrolyte chain will collapse to adopt the conformation of a neutral chain in good solvent (Scheme 4b).
1.2.2 Manning Condensation and Effective Charge Density
The properties of polyelectrolyte solutions depend strongly on the interactions between the polymers and the surrounding counterions. Manning’s theory of counterion condensation predicts that a certain quantity of counterions condenses onto a polymer, whose charge density exceeds a critical value [44]. This leads to an effective decrease in the polymer charge. The macroscopic properties of the polyelectrolyte are not determined by its bare charge but by an effective charge. In particular, the flexibility and hydrophobicity of the polyelectrolyte chain, the chemical nature of the counterions, the solvent quality, and concentration effects may well influence the “Manning condensation” [45]. Condensation occurs whenever the average distance between co-ions (assumed to be monovalent) on the polymer backbone is smaller than the Bjerrum length λ B (distance between two dissociated ion pairs) defined as:
where q is the elementary charge, k B T the thermal energy, and ε the dielectric constant of the solvent. This condensation is expected to lead to an average charge density of q/λ B on the polymer backbone.
Since the polyelectrolyte dissociation releases counter-ions, this affects the solution’s ionic strength and consequently the Debye length (distance over which significant charge separation can occur). The Debye length κ −1 (in nm) can be expressed as:
where N A is the Avogadro number, λ B is the Bjerrum length of the medium (in nm), and I is the ionic strength of the medium (in mol L−1).
At room temperature, in water, the relation gives [46]:
These are parameters that should be taken into account when considering the individual polyelectrolytes (DNA and polycations) before complexation.
1.3 DNA/Polycation Complexes
Polyanions and polycations can co-react in aqueous solution to form polyelectrolyte complexes via a process closely linked to self-assembly processes [47]. Despite progresses in the field of (inter-) polyelectrolyte complexes [47] (IPEC from Gohy et al. [48], block ionomer complexes BIC from Kabanov et al. [49], polyion complex PIC from Kataoka and colleagues [50, 51], and complex coacervate core micelles C3M from Cohen Stuart and colleagues [52], understanding of more complex structures such as polyplexes (polyelectrolyte complexes of DNA and polycations) [53] is rather limited [54]. It has also to be considered that the behavior of cationic polymers in the presence of DNA and their complexes can be unpredictable, particularly in physiological environments due to the presence of other polyelectrolytes (i.e., proteins and enzymes) and variations in pH, etc.
1.3.1 Factors Influencing the Complexation of DNA by Cationic Polymers
The complexation of DNA and polycations is a function of the intrinsic properties of the two components. For instance, from the use of synthetic polycations for complexing DNA also arises the problem of polydispersity of polymers (a polymer sample is usually composed of macromolecular species of differing molar masses) compared with DNA, which is monodisperse. Because the polydispersity of the polycation could be an issue in studies of IPECs, sugar-based polymers (usually polydisperse except if fractionated), conjugated polymers (polydispersity, M w/M n > 2), branched PEI derivatives, and hyperbranched polymers are out of the scope of this review, as already mentioned. Only polymers synthesized via controlled or living polymerization methods will be discussed [55–57].
Although the interaction between multivalent polymeric cations with DNA is electrostatic in origin, the flexibility of the polymer backbones (rigid versus flexible) and molecular architectures also show great impact on the properties of the final polyplexes [58]. The molecular weight and topology of both the polymer (which can possess various architectures such as linear, brush, star, etc.) and the DNA (linear, circular, and supercoiled) has to be taken into account. On the polymer side, the composition (block, statistical, random, etc.) and its strength as a polyelectrolyte also play a role, as its charge density is varied.
As already mentioned, the main driving force of complex formation is the gain in entropy caused by the release of low molecular weight counterions, but other interactions such as hydrogen bonding and hydrophobic interactions can also contribute to the complexation process. Thus, the hydrophilicity/hydrophobicity of the polymer (influencing both the solubility of the polymer in aqueous media and its complexation with DNA via hydrophobic interactions) as well as its H-bonding capacity have to be taken into consideration. Moreover, the importance of counterions or substituents (inducing screening of charges) is often neglected in the formation of polyplexes.
Extrinsic factors (environment) such as the medium conditions also play a large part in the complexation process, especially pH and ionic strength (salt and polyelectrolyte concentrations). Also of prime importance is the way that the complexation itself is conducted, i.e., mixing parameters such as the stoichiometry of the components, the addition rate, and order of addition of the components (kinetic versus thermodynamic). Even if this process is fast and kinetically controlled (in water without added salt), i.e., far from the thermodynamic equilibrium, it can be followed by a slower stage in which the chains redistribute to a IPEC conformation closer to equilibrium [59].
1.3.2 Condensation of DNA by Cationic Polymers
DNA can be more simply considered as a particular case of a stiff anionic linear polyelectrolyte. Monovalent cations will condense on DNA (condensation) but do not cause DNA compaction, which is the collapse of DNA into a compact structure. The compaction of DNA by an incompatible polymer has been modeled as a coil–globule transition such as observed in other polymers [60], and is also the topic of recent studies by the group of Dias, Lindman and colleagues [61, 62].
What seems to be the predominant method for polyplex formation is the addition of a polymer solution to a DNA solution. Some of the consequences of this procedure are that the concentration of the DNA solution changes in course of the addition (increase in volume) and DNA is consumed by the ongoing complexation process. Despite the importance of the addition rate, it is often not mentioned in polyplex studies. For instance, from IPEC studies it was found that the higher the titrant addition rate, the higher the storage stability of the complexes in the case of random copolymers of sodium 2-acrylamido-2-methylpropanesulfonate with either t-butyl acrylamide or methyl methacrylate complexed with poly(diallyldimethylammonium chloride) or with an ionene-type polycation containing 95 mol% N,N-dimethyl-2-hydroxypropyleneammonium chloride repeat units [63]. Moreover, by addition of a polycation to DNA, the zeta potential increases from negative values (DNA) to positive values (nanoparticles with excess of polycations).
The behavior of both DNA and polyplexes is also a function of the starting concentration of DNA, which can be in the dilute (polymers act as individual units without intermolecular interactions), intermediate, or semi-dilute regime (polymer chains overlap each other and form a transient network). IPEC studies of the complexation of poly(allylamine hydrochloride) and the two polyanions poly(acrylic acid) and poly(methacrylic acid) have shown that the higher the concentration, the larger and denser are the complexes formed [64]. Unfortunately, this type of study with complexes of DNA and polycations are still scarce.
1.3.2.1 Structural Models
Two structural models are discussed in the literature for polyelectrolyte complex (PEC) formation, depending on the components (weak or strong polyelectrolyte, stoichiometry, molecular weight) and the external conditions (presence of salts, etc.): ladder-like (complex formation takes place on a molecular level via conformational adaptation) or “scrambled egg” structure (large number of chains in a particle) (Scheme 5) [65].

Representation of ladder and scrambled egg structures. Black lines represent large polyions (negative), while gray lines with squares represent polyions of opposite charge (positive). (a) Ladder representation, where insufficient ion pairing occurs under certain stoichiometric conditions leading to macromolecular aggregates, insoluble, and soluble PECs. (b) Scrambled egg model, where polymers of comparable size complex to yield insoluble PECs under certain conditions. Reprinted with permission from [65]. Copyright 2007 Springer
The ladder-like structure results from the mixing of polyelectrolytes having weak ionic groups and large differences in molecular dimensions. It is the result of the propagation of the complex reaction as a “zippering action,” since the ionic sites next to the first reacted ones would be the most likely to react next. The “scrambled egg” structure refers to complexes that are the product of the combination of polyelectrolytes having strong ionic groups and comparable molecular dimensions. These models have been extensively discussed and most experimental structures lie between these two models, though probably closer to the scrambled egg than the ladder model [66], especially in the case of complexes of DNA with polycations.
1.3.2.2 Cooperative Versus Non-cooperative Binding
The binding itself can occur either via cooperative or non-cooperative binding (Scheme 6). A macromolecule (DNA, protein, synthetic polymer) exhibits cooperative binding if its affinity for its ligand changes with the amount of ligand already bound. The cooperativity is positive if the binding of ligand at one site increases the affinity for ligand at another site, whereas the cooperativity is negative if the binding of ligand at one site lowers the affinity for ligand at another site. A macromolecule exhibits non-cooperative binding if the ligand binds at each site independently.

(a) If a polycation binds to a cluster of DNA binding sites in a non-cooperative manner, a gradual increase in polycation concentration generates a gradual increase in the average occupancy of the cluster. (b) Conversely, if the polycation binding to adjacent sites is cooperative, a gradual increase in polycation concentration generates a “digital” on/off response as the concentration sweeps a threshold value (dashed line). The higher the binding cooperativity, the steeper the transition between the “off” and the “on” states. Adapted and reprinted with permission from [67]. Copyright 2010 Elsevier
In complexation of DNA with polycations, both scenarios can be found. In the case of cooperative binding, some of the DNA is totally complexed, while the rest of DNA is left “naked.” In the case of non-cooperative binding, all individual DNA chains are roughly equally complexed by polycations.
1.3.3 Structure of Polyplexes
Condensates of polycation with DNA (polyplexes) can adopt various shapes, the most commonly observed being toroidal, rod-like, and globular (examples of some of these structures are presented in Scheme 7) [68–71]. The different structures that IPECs can adopt can be categorized into different subtypes: water-soluble, colloidally stable, and insoluble. The type of complex formed is governed by all the factors mentioned in the previous paragraphs. Moreover, it should be noted that the polycation/DNA charge ratio influences the size, charge, and solubility of the polyplexes. As a consequence, in some cases, the polyplexes can be consecutively water soluble, then colloidally stable, and eventually precipitate.

Tapping mode AFM height topographs of (A) uncomplexed pBR322, (C) linear DNA, and (B, D) the respective complexes of these formed when mixed with chitosan. C DNA = 4μg mL−1. Reprinted with permission from [68]. Copyright 2004 American Chemical Society
In the schemes included hereafter DNA is represented as linear (but can have other topologies and is compacted within the polyplexes), and the type of binding (cooperative or not) is not taken into account, except if mentioned.
1.3.3.1 Water-Soluble Polyplexes
Soluble polyplexes are macroscopically homogeneous systems containing small PEC aggregates. Due to the strong polyanionic nature of DNA, water-soluble polyplexes are formed when the polycation is present in non-stoichiometric proportions under certain concentration (dilute) and/or salt conditions and with significantly different molecular weights [72, 73]. The complex adopts a conformation similar to that of the ladder model with hydrophilic (polyanion) and hydrophobic segments (complex of polyanion and polycation) (Scheme 8). This type of polyplexes is the key to forming polyplexes monomolecular in one component.

Proposed model for water-soluble polyplexes with sequentially hydrophilic (polyanion) and hydrophobic segments (complex of polyanion and polycation). The bending or compaction of DNA are not taken into account in this scheme
1.3.3.2 Colloidally Stable Polyplexes
Colloidally stable polyplexes are PEC systems in the transition range to phase separation, exhibiting an observable light scattering or Tyndall effect [65]. These systems can be stable because of electrostatic stabilization, steric stabilization, or a combination of both called electrosteric stabilization.
1.3.3.2.1 Without Steric Stabilization
IPEC formation between (strong) polyelectrolytes results in highly aggregated and/or macroscopic flocculated systems. Nevertheless, the aggregation can be stopped at a colloidal level in extremely dilute solutions, and a polydisperse colloidally stable system of nearly spherical particles can usually be achieved [47].
Due to the entropy-driven charge neutralization rather than a strictly located binding, charges can sometimes be “buried,” leading to a mismatching of the charge densities even at 1:1 charge ratio. Furthermore, the stoichiometry depends on the polymer flexibility because rigid polymers that are less able to change their conformation are more likely to form non-stoichiometric IPECs. Polymers with nonlinear architectures (graft, hyperbranched, etc.) are also prone to the formation of non-stoichiometric IPECs, since charges at sites in the inner parts of the molecules can be inaccessible to the oppositely charged polyelectrolyte. Mismatching charge densities leads to a higher degree of swelling of the colloidal particles. It is proposed that these colloidally stable IPEC particles consist of a charge-neutralized core, in which 1:1 stoichiometry and high entanglements prevails, and an outer shell consisting of a few polyelectrolyte layers whose charges are not completely compensated, giving the complex its net charge, stabilizing the particles, and preventing them from further aggregation. The number of polymer chains included in a single IPEC particle varies from hundreds of chains in extremely dilute systems up to several thousands for more concentrated component solutions [47].
1.3.3.2.2 With Steric Stabilization
In the previous situation, the particles were stabilized mainly by the charges in the outer shell (electrostatic stabilization). Another type of stabilization for colloids is steric stabilization, which can be introduced in the case of polyplexes by the presence of a neutral hydrophilic block in the polycation. Micelle-like structures are thus obtained consisting of a charge-neutralized core, in which 1:1 stoichiometry and high entanglements prevails, and an outer shell consisting of a neutral hydrophilic block, stabilizing the particles via steric interactions. These IPEC micelles are also called complex coacervate core micelles (CCCM or C3M) [52]. This allows, even at charge neutralization and despite possible secondary aggregation, the colloids to stay in solution stabilized by their polymeric hydrophilic shell. Secondary aggregation occurs when the particles in solution try to minimize contact with their surroundings (water) at charge neutralization; the particles will adhere with each other and finally the entire dispersion may coalesce. Usually, the higher the molecular weight of the polymer and the larger the thickness of its hydration shell, the more stable are the colloids. In the most efficient cases of steric stabilization, secondary aggregation can be avoided and single particles are present in solution, even if neutral. If the stabilization is slightly less efficient, the aggregates that are nevertheless stable can be redispersed by the addition of more polycation. The additional polymer is included in the polyplexes leading to a positive net charge, which introduces repulsion between the particles (Scheme 9).

Proposed model for sterically stabilized polyplexes as function of the charge ratio polycation:DNA. When an excess of DNA is present in solution, if the binding is cooperative then neutral polyplexes (charge neutralized DNA/polymer complexes) and DNA molecules will coexist in solution. If the binding is not cooperative, negatively charged polyplexes will be present in solution (where the charges of DNA are not compensated by the polycations). In both cases negative zeta potentials are obtained. At charge neutralization, if the steric stabilization is not sufficient, aggregation of the neutral polyplexes will take place and they will precipitate (they can eventually in some cases be redispersed following further addition of polymer).If the steric stabilization is sufficient, polyplexes can stay as individual nanoparticles in solution. When an excess of polymer is present in solution, two cases are possible: either the polycations and neutral polyplexes coexist in solution because the polycations do not adsorb at the surface of the polyplexes, or the polycations adsorb on the polyplexes surfaces (usually when the steric barrier is sufficient) leading to positively charged polyplexes (until a certain point where the polycations do not adsorb on the positively charged polyplexes due to electrostatic repulsion). In both cases positive zeta potentials are obtained
Poly(ethylene glycol) (PEG) is the polymer that is most used for steric stabilization due to its biocompatibility. It should be noted that random copolymers are usually not as effective in steric stabilization as block or graft copolymers.
In most of the studies, unfortunately, physico-chemical characterization is not conducted in enough detail that the size and surface charge of the various species present in solution are determined; usually, only the properties of the colloidal suspensions (sum of species) are determined. Indeed, when polycations are added in high excess to polyplexes after charge neutralization, it seems that in most cases polycations and neutral polyplexes coexist in solution because the polycations do not adsorb at the surface of the polyplexes. A way of determining the real size and surface charge of the polyplexes would be to separate the colloids from the individual polycations, for instance by means of dialysis (dilution could eventually have an influence on the stability of the polyplexes by influencing the equilibrium between polycation and PIC micelle). In both cases (excess polycation adsorbed or not at the surface of the polyplex), this would explain the big discrepancies between the physico-chemical characteristics of polyplexes and their performances in vitro. Even if the mixture of polymer and polyplex shows a positive zeta potential, this does not mean that the polyplex containing the therapeutic gene is positively charged. As a consequence, if the polyplex itself is neutral, it will not interact favorably with the cell membrane and thus will not lead to high transfection because of the low cellular uptake. Also, even if the polyplex has a positive zeta potential due to the polycations adsorbed on the surface of the neutral polyplexes, the polycation would probably be easily displaced after intravenous injection, i.e., in vivo (dilution effect and competition with other polyanions or polycations present in the blood stream).
1.3.3.3 Insoluble Polyplexes
Insoluble polyplexes are the result of a two-phase system of supernatant liquid solution (containing either polycation as in Scheme 10, or DNA as in Scheme 11) or colloidal suspension (in the particular case of polymeric micelles in solution as shown in Scheme 11, case 1) from which the polyplexes precipitate. For non-sterically stabilized polyplexes, when the molar mixing ratio approaches unity or a low quantity of salt is added (screening of charges of the polyplex) [74] and therefore the electrostatic stabilization is insufficient, secondary aggregation occurs and the aggregates precipitate (this is also a function of the density of the polyplexes) (Scheme 10) [47].

Proposed model for non-sterically stabilized polyplexes as function of the charge ratio strong polycation:DNA. When an excess of DNA is present in solution, if the binding is cooperative neutral polyplexes (charge neutralized DNA/polymer complexes) and DNA molecules will coexist in solution. If the binding is not cooperative, negatively charged polyplexes will be present in solution (where the charges of DNA are not yet compensated by the polycations). In both cases negative zeta potentials are obtained. At charge neutralization, aggregation of the neutral polyplexes will take place and they will precipitate (macroscopically visible). These aggregates of polyplexes cannot be redispersed and addition of more polymer will lead to positive zeta potential because the polycation is the only specie present in solution

Proposed models for polyplexes based on amphiphilic polycations as a function of the charge ratio. Case 1: When an excess of DNA is present in solution, if the binding is cooperative neutral polyplexes with hydrophobic shell and DNA molecules will coexist in solution. As these core–shell structures possess hydrophobic shells, their range of stability is reduced and they nearly immediately aggregate and precipitate. If the binding is not cooperative, negatively charged polyplexes with hydrophobic shell will be present in solution (where the charges of DNA are not yet compensated by the polycations). At charge neutralization, aggregation of the neutral polyplexes will take place and they will precipitate. With further addition of polymer, amphiphilic polycations are present in solution as unimers until the CMC is reached, where polycationic micelles are the only colloidal specie in solution. Case 2: When DNA is added to a polymer micellar solution, if the binding is cooperative neutral polyplexes with a micellar core composed of the cationic amphiphilic and a shell composed of DNA are formed. If the binding is not cooperative, overall positively charged polyplexes with DNA as shell will be present in solution. At charge neutralization, aggregation of the neutral polyplexes will take place and they will precipitate. With further addition of DNA, the zeta potential of the solution will be negative because DNA is the only specie present in solution
1.3.3.4 Polyplexes Based on Amphiphilic Polycations
Polymeric micelles are colloidal particles formed by the self-assembly of amphiphilic block polymers (at certain hydrophilic/hydrophobic ratio of the blocks constituting the polymer) in an aqueous environment and have sizes ranging from 10 to 100 nm [75]. Micelles can form only above a given concentration, which is known as the critical micelle concentration (CMC) [76]. If the concentration of amphiphilic polymer in the sample is under its CMC, the observed behavior is roughly that presented in previous cases, except that, due to the amphiphilic nature of the polycation, the stability of the polyplex will be more limited in an aqueous environment.
If the concentration of the amphiphilic polymer in the sample is above its CMC, there are two cases to consider: the micellar solution is added to the DNA solution or the DNA solution is added to a micellar solution. In the first case, where the micellar solution of polycation is added to DNA, the micelles are immediately diluted in the DNA solution, which on one hand can mean that the polymer concentration is under its CMC and therefore that the polymer is only present as individual chains in solution (unimers), on the other hand, that there is concurrence between self-assembly of micelles versus electrostatic interactions with a large quantity of negatively charged material. In this case, the micelles are usually destabilized as soon as they reach the DNA solution (Scheme 11, case 1).
In the case of DNA added to a micellar solution of polycation, the micelles can stay stable in some cases: (it depends on the hydrophobicity of the micellar core and strength of the electrostatic interactions). This can be proven if pyrene or other hydrophobic molecules entrapped in the hydrophobic interior are not released even after addition of DNA [77]. It has to be noted that micelles are in thermodynamic equilibrium with unimers and that both species can form electrostatic interactions with DNA. When the micelles do not undergo a structural change, no rearrangement into a “scrambled eggs” structure takes place between the amphiphilic polycation and DNA, and because DNA is in minority, it adds to the positive shell of the structure until neutralization (Scheme 11, case 2).
1.3.3.5 Influence of Salts
After changes in ionic strength (due to the addition of salt), swelling or deswelling of IPECs occurs immediately, whereas coagulation (i.e., destabilization of colloids by neutralizing the electric charge of the dispersed nanoparticles, which results in aggregation of the colloidal particles) is a much slower process and is dependent on the concentration of the colloidal particles [74]. Two major effects on the formation of IPECs in the presence of salt were found by Dautzenberg [77]. On the one hand, the presence of a very small amount of salt during formation dramatically decreased the level of aggregation, probably due to the less stiff and more coiled structure that the polymers can adopt. On the other hand, a higher ionic strength resulted in macroscopic flocculation, explained by the contribution of two factors: particle swelling because of charge screening of the stabilizing outer shell and particle aggregation due to colloidal instability. However, the internal structure of most IPECs is marginally affected by salt [77]. With a further increase in ionic strength, the point is reached where charges are screened at the level of the polymers, and polycations and DNA are dissolved as individual polymers.
As already mentioned, counterions seem to be important for the interaction between polyelectrolytes (uni- or multivalent) and the specific ions involved (size, chaotropic/kosmotropic) [78, 79].
1.3.4 Characterization of Polyplexes
The final polyplexes either precipitate if they are water insoluble or form a colloidal system (particles that have a diameter less than a micrometer evenly dispersed in aqueous media in this case) if they are sufficiently stabilized even after complexation. In both cases, these polymer–DNA complexes can be characterized using a variety of analytical techniques, which will be presented in the next paragraph.
In the case that the polyplexes form a colloidal system, the Derjaguin, Landau, Verwey and Overbeek theory (DLVO theory) can be used to describe their colloidal stability and aggregation behavior. The DLVO theory describes the force between charged surfaces interacting through a liquid. It takes into account the effects of the van der Waals attraction and the electrostatic repulsion due to the double layer of counterions, but additional forces have also been reported to play a major role in determining colloid stability (Fig. 1) [80]. This topic is addressed in more detail by Lebovka in another chapter of this volume [81].

Interactions between nanoparticles. (a) Traditional forces for colloidal stabilization (e.g., electrostatic, van der Waals, steric) that occur when particles are dispersed in aqueous media. (b) The van der Waals forces are attractive whereas the electrostatic forces are repulsive over a typical length scale. The Derjaguin–Landau–Verwey–Overbeek theory in colloid science considers the sum of these forces. Reprinted with permission from [80]. Copyright 2011 Elsevier
1.3.4.1 Structural Characterization
Light scattering (LS) provides information related to the dimensions of the polyplexes (hydrodynamic radius R h), their shape (radius of gyration R g and shape factor ρ = R g/R h), as well as weight-average molecular weight (M w) of the aggregates and polydispersity of the sample.
Atomic force microscopy (AFM) and electron microscopy techniques allow imaging the polyplexes. The electron microscopy techniques used, as for other nanoparticles, are transmission electron microscopy (TEM) and cryo-TEM.
1.3.4.2 Charge Determination
In the case of strong polyelectrolytes, the number of ionized units corresponds to the number of dissociable ionic units (see Manning condensation) and is independent of the pH. For weak polyelectrolytes, the number of ionized units at a given pH is dependent on the pK a. From acid/base titration, their pK a as well as buffering capacity (illustrated by plotting the pH of a solution containing a polymer as a function of the volume of acid added) can be determined. The following equation reported by Patchornik et al. can be used to determine the number of ionized units, i.e., the protonation state of a polycation, at a specific pH [82]:
where pK a is the intrinsic pK of the protonatable moiety, n is the average number of protonatable moieties per polymer chain, α is the fraction of protonated moieties, and w is an electrostatic interaction factor defined as:
assuming a spherical molecule with radius b and a distance of closest approach a; D is the dielectric constant of water, k the Boltzmann constant, T the absolute temperature, e the electronic charge and k has its usual significance in the Debye theory. By solving it in an iterative fashion, one can determine the percentage of groups on the polymer that are protonated at physiological pH and therefore are potentially available to assist in the condensation of DNA.
Z or φ is the charge ratio at a given pH (also called +/−), meaning the ratio of ionized units of the cationic polymers at the given pH by the number of negative charges of the DNA. N:P ratio, which is the ratio of nitrogen atoms in the polycation to phosphorus atoms in DNA, is usually employed in the case of polyplexes based on weak polyelectrolytes when the number of ionized units is not determined. Unfortunately, it does not best reflect the polyelectrolyte behavior. Also used are the molar or weight ratios of polymer:DNA.
Zeta potential (ζ) analysis can be used to measure the relative surface charges of nanoparticles such as polyplexes. It helps define a range of stability for colloids, when steric stabilization does not take place (only electrostatic stabilization). Zeta potential, as well as dynamic light scattering (DLS) are useful methods for determining if various fractions are present in solution (with different surface charge or size, respectively).
Agarose gel electrophoresis separates macromolecules on the basis of both charge and size, and the immobilization of DNA on a gel in the presence of cationic polymer can be used to determine the conditions under which self-assembly and/or charge neutralization occurs. It should be noted that retardation of polyplexes can be due to neutralization of the positive charge or to an increase in mass.
1.3.4.3 Strength of the Complexation
Ethidium bromide (EtBr) is commonly used as a fluorescent nucleic acid stain in techniques such as agarose gel electrophoresis. When excited by 530 nm light, EtBr emits fluorescence at 610 nm, with an almost 20-fold increase in intensity after intercalating into DNA base pairs due to π-stacking with the nucleobases (see Fig. 2b for an example of a DNA intercalator) [84]. When polymers interact tightly with DNA to form polyplexes and condense DNA, EtBr is released into solution, where its fluorescence is far inferior to that when intercalated in DNA. Thus, EtBr is a good indicator for evaluating the strength of condensation of DNA by polycations. Some other dyes such as Hoechst stains are (minor) groove binding agents (see Fig. 2a), and therefore give less information about the strength of complexation.

(a) Groove binding of Hoechst 33258 to the minor groove of DNA. (b) Intercalation of ellipticine into DNA. Adapted and reprinted with permission from [83]. Copyright 2007 Elsevier
The coil–globule transition of DNA (reflecting the compaction of DNA) can be followed by thermal analysis or spectroscopic methods such as UV.
Competition binding can be used to test the stability of the polyplexes. The release rate of DNA from a polyplex by competitive binding between the components of the gene delivery vector with charged components (for instance polyanions such as heparin) is an indicator of the strength of the complexation (necessary for the extracellular milieu) as well as the possibility of release (in intracellular milieu, favorable for gene expression).
Testing the efficiency of a polymer in protecting DNA from enzymatic degradation (by nuclease, etc.) gives information about the efficiency of compaction of DNA by the polycation and/or the steric protection of the polyplex. The protection of DNA in the polyplex from its degradation by enzymes is essential for in vivo delivery.
1.4 Application in Gene Therapy
One of the many applications of polymers capable of complexing but also condensing DNA is their use as transfection agents (introduce genetic material into cells).
1.4.1 Introduction to Gene Therapy
Gene therapy aims to cure inherited and acquired diseases by correcting the overexpression or underexpression of defective genes, and its success depends largely upon the development of vectors that deliver and efficiently express a therapeutic gene in a specific cell population [85, 86]. Gene therapy protocols were originally designed to correct inheritable disorders such as adenosine deaminase deficiency, cystic fibrosis, Gaucher’s disease, and Duchenne muscular dystrophy [87, 88]. However, gene therapy is not exclusively used in an attempt to supply a missing gene product to a patient with a given inborn error of metabolism. Indeed, gene therapy has been considered more recently as a promising tool for treating acquired diseases such as cancer [89] and human immunodeficiency virus (HIV) infections [90]. Clearly, different applications have distinct needs, and tailoring gene delivery vectors to the specific requirements of a therapeutic application is still a challenge. For example, the ideal gene vector for treating genetic disorders should not only deliver intact pDNA efficiently to the nucleus of most of the target cells, but also, once delivered, the transgene should be maintained in the nucleus without disrupting host gene expression or signaling pathways. By contrast, anti-cancer gene therapy trials are in progress in which the aim is high transgene expression in as many tumor cells as possible, rather than sustained gene expression.
Two types of vectors are used in gene delivery: viral [91] and non-viral [92, 93]. Viral-mediated DNA vehicles (infection) have played a major role in gene therapeutics. Unfortunately, the initial enthusiasm associated with the high infection yields has been tempered by growing concerns regarding safety issues such as toxicity, immunogenicity, and oncogenicity. On the other hand, synthetic gene vectors (transfection), with dimensions in the nanometer range, provide potential alternatives for gene therapy because these vectors (based on lipids, dendrimers, peptides, or polymers) are more easily produced and at lower cost. Moreover, they work reasonably well in vitro and overcome some of the disadvantages of viral-based gene delivery systems such as immunological response, fatal infections, etc.
The genetic material for treatment of a variety of genetic disorders can be of three types: (1) pDNA, to express a gene of interest under the control of a suitable promoter (has to reach the nucleus), which will result in the increased production of a protein [94, 95]; (2) oligomeric genetic material such as antisense OligoDeoxyNucleotides (ODN), short RNA molecules such as small interfering RNA (siRNA) micro-RNA (miRNA) or short hairpin RNA (shRNA), or a DNAzyme in order to silence a specific gene by reducing the target/protein activity. The short RNA molecules as well as DNAzyme have to reach the cytoplasm and more precisely the RNA-induced silencing complex (RISC) without being destroyed in the late endosomes or lysosomes [96–98].
1.4.2 Requirements for Efficient Gene Therapy
1.4.2.1 Complexation and Compaction/Condensation
For most cell types, the size requirement for particle uptake via endocytosis is of the order of 200 nm or less. As DNA has a R h of a few hundred nanometers when its molecular weight is a few thousand base pairs, the polymers should not only complex DNA but also condense or compact it into smaller particles. The polymer remains unable to condense DNA until the neutralization of a critical amount of negative charges on the DNA. For instance, Wilson and Bloomfield have calculated that in order to condense DNA, 90% of the phosphonate moieties have to be neutralized when the condensing agent is spermine or spermidine [99].
For condensation, strong (quaternary ammonium, etc.) as well as weak polyelectrolytes (containing amino acids such as Arg, Lys, etc. or poly[(2-dimethylamino) ethyl methacrylate], PDMAEMA [100]) can be used, which should possess a minimum number of cationic charges at physiological pH. For instance, as a weak polyelectrolyte, linear PEI (LPEI, 22 kDa) has 75% of its amino groups protonated at physiological pH [101]. At pH 8, with a degree of polymerization (DP) of 32, PDMAEMA has approximately 24% of its amino groups protonated [102].
1.4.2.2 Extracellular Barriers and Physico-chemical Aspects
A major drawback of current transfection vectors is that they have poor in vivo transfection efficiency and only confer transient gene expression. Indeed, poor transfection efficiency is due, in part, to the lack of stability of the non-viral vector–DNA complex under physiological conditions and its ability to interact with blood plasma proteins after intravenous injection, the extracellular matrix, and undesirable cells, and its possible degradation by enzymes, even before reaching the intracellular compartment (Scheme 12). In order to overcome these problems and to enable the carrier to translocate across cellular membranes (thus further influencing its biodistribution, cell internalization, and trafficking properties), it is of prime importance to engineer the polyplex (polymer/DNA complex) in terms of chemical structure, molecular weight, hydrophilicity/hydrophobicity, size, surface groups, charge density, and concentration. This is the main aspect that will be treated in this review, i.e., the chemical engineering of polycations and the physico-chemical aspects of polyplexes. Various publications address the topic of structure–property relationships by modification of the polymer via processes such as acetylation [103, 104], introduction of an hydrophilic block through PEGylation [105, 106], control of charge density [102, 107], incorporation of hydrogen bonding [107, 108], or varying the topology of the polymer [109, 110]. It is important to note that internalization of positively charged polyplexes is facilitated, given that the cell surface is negatively charged (because of the presence of proteoglycans) so, in general, nanoparticles with smaller size and higher zeta potential are most likely to be uptaken by cells.

Nucleic acid nanoparticle formation and delivery barriers. Adapted and reprinted with permission from [95]. Copyright 2012 Elsevier
Once in the blood stream, hydrophobic nanospheres are rapidly opsonized and extensively cleared by the mononuclear phagocyte system (MPS). This problem can be prevented by surface modification, such as coating with hydrophilic polymers, or by formulating nanospheres with biodegradable copolymers with hydrophilic characteristics [111]. Moreover, the introduction of hydrophilic polymers, such as oligo(ethylene glycol) or poly(ethylene glycol) (PEG, probably the most widely used), or others such as the zwiterrionic 2-methacryloxyethyl phosphorylcholine (MPC) or poly(hydroxyethyl methacrylate) (PHEMA) can provide steric stabilization to otherwise unstable polyplexes in water. They can also protect DNA against protein adsorption and degradation by enzymatic nucleases.
Active targeting to certain cells or organs can be attained by the recognition at the molecular level between a ligand and receptors overexpressed on cell membranes through specific interactions. Once the molecules bind to the receptors, the complex is internalized via receptor-mediated endocytosis, facilitating the cellular uptake of the carrier of this ligand. This will not be treated in this review because, most of the time, active targeting with nanoparticles is achieved by conjugating an antibody or protein to a polymer, thus influencing the physico-chemical properties of the polymer and polyplex formed thereof. Nevertheless, this is an extremely important aspect of gene delivery for in vivo applications [112].
1.4.2.3 Intracellular Processes
As previously mentioned, for most cell types, the size requirement for particle uptake via endocytosis is in the order of 200 nm or less, and a net positive charge on the surface of the conjugate has been shown to be important for triggering uptake. Moreover, to be effective, these polyplexes must be optimized at all stages of the delivery process, ranging from target-cell recognition (attachment of targeting ligands in order to be recognized and taken up by specific cells) [113–115] to their escape from the endosome-enclosed milieu, resistance to cytoplasmic degradative enzymes such as nucleases, and release of the genetic material at the desired site of action [116]. Thus, polymers should bind efficiently and protect the genetic material against nonspecific interactions with proteins and cell membranes in blood, but efficiently release it in the cytosol in order to favor gene expression (Scheme 13) [117]. Indeed, when the polycation binds too strongly, it results in impaired gene expression.

Intracellular trafficking of polyplexes. The size of a polyplex is generally a few hundred nanometers (100–200 nm). Reprinted with permission from [100]. Copyright 2012 Elsevier
Concerning the intracellular trafficking of polyplexes, it begins in early endosomal vesicles. These early endosomes subsequently fuse with sorting endosomes, which in turn transfer their contents to the late endosomes. Late endosomal vesicles are acidified (pH 5–6) by membrane-bound proton-pump ATPases. The normal process is that the endosomal content is then relocated to the lysosomes, which are further acidified (pH ~4.5) and contain various nucleases that promote the degradation of the DNA. To avoid lysosomal degradation, the genetic material (free or complexed with the carrier) must escape from the endosome into the cytosol (endosomal escape, Scheme 13) [118].
Release into the cytosol can be achieved by using bioresponsive polymers for triggered release (responsive to conditions or components present in the intracellular milieu) [118]. This type of polymer, such as those containing disulfide bonds, will not be discussed here because they are of little relevance to polyelectrolyte interactions and DNA complexation. Another way to favor endosomal escape is to use polymers that have a pH-buffering effect or “proton sponge effect.” These polymers must contain amines that can act as a “proton sponge” in endosomes, preventing acidification of endosomal vesicles and thereby increasing the ATPase-mediated influx of protons and counter-ions (which enter the vesicles to balance the proton flux), leading to osmotic swelling, endosomal membrane rupture, and the eventual leakage of the polyplex into the cytosol (Scheme 14) [120].

The proposed proton sponge mechanism of endosomal escape. (A) Polyplexes enclosed in an endosome after endocytosis. (B) Due to the pH buffering in the endosome, the protons continue to be pumped into the vesicle, resulting in Cl− influx and an increase in the osmolarity inside the endosomal vesicle. (C) Because of the osmolarity increase, water passes into the endosomal vesicle. (D) The increase in water volume results in the swelling of the endosomal compartment until it ruptures. (E) Release of the polyplex into the cytoplasm, which leads to nuclear uptake of DNA. Reprinted with permission from [119]. Copyright 2011 Elsevier
1.4.2.4 Toxicity, Biocompatibility, and Biodegradability
The challenge is not limited to bringing the polyplex inside cells: even if the polyplexes overcome the extracellular barriers, it is not useful if, due do its intrinsic toxicity, the polyplex kills cells after uptake. This is in many cases the reason why the overall transfection efficiency of a polyplex is rather low, despite a high value for its cellular uptake. Thus, it is of prime importance to study the intracellular uptake, for example with fluorescence imaging, as well as the toxicity of both the polymer and polyplex and the transfection efficiency of the polyplex at relevant concentrations and times of exposure. Almost as important is the comparison with a relevant standard such as PEI, because usually more than one parameter is varied (cell line, concentration, etc.) from one study to another and therefore the results are difficult to compare. In toxicity tests, the half maximal inhibitory concentration or IC50 is the quantitative measure used (in some of the publications IC50 also means the charge ratio causing 50% reduction of EtBr fluorescence). The cell lines most commonly used are: COS cell lines (CV-1, simian in origin, and carrying the SV40 genetic material), which resemble human fibroblast cells; human embryonic kidney 293 cell line (HEK 293), which is originally derived from human embryonic kidney cells grown in tissue culture; and HepG2 cell line that is a human liver hepatocellular carcinoma cell line.
It is to be mentioned that, as a general rule, strong polycations are highly toxic [121]. It is nevertheless possible to limit immediate toxicity by “masking” the non-biocompatible part to its environment via the introduction of hydrophilic biocompatible segments (such as PEG) into the construct. Biocompatibility is the ability of a polymer or material to perform with an appropriate host response (local and systemic) in a specific application and by not producing a toxic, injurious, or immunological response in living tissue. This is strongly determined by the primary chemical structure. Sugar-based polymers (chitosan, dextran, etc.) are a great example of biocompatible materials. Increasing the biodegradability is of course an alternative way to limit toxicity (for instance, by incorporation of acid labile groups such as β-amino esters and ortho esters) because the byproducts of degradation can be eliminated by the body via natural pathways.
It is important to keep following criteria in mind for efficient polymer-mediated gene delivery: efficient compaction of genetic material (size <200 nm); stability of the polyplexes under physiological conditions (i.e., presence of salts, pH 7.4) because particles that precipitate under these conditions are not suitable for in vivo applications; high uptake by cells and intracellular release; and efficient transfection without inducing cytotoxicity. Moreover, biodegradability and targeting of the polyplexes are important properties for in vivo applications.
2 Polycation/DNA Complexes
2.1 Water-Soluble Polycations
2.1.1 Strong Polyelectrolytes
Strong polyelectrolytes are salts of quaternary ammonium cations (alkyl), pyridinium, or imidazolium with an anion. Charge neutralization is usually achieved at or close to a 1:1 stoichiometry for strong polycations and DNA.
2.1.1.1 Containing Aromatics or Having a Charged Backbone
In the pioneering work of Izumrudov and Zhiryakova, the shorter the PEVP (Fig. 3a) the less resistant was the polyplex to the addition of salts; this effect was much more pronounced for chain lengths between 10 and 100 than above [122]. Interestingly, the stability of the PEC was virtually independent of the length of the nucleic acid in the studied region (500 bp, DNA from salmon testes; 10,000 bp, calf thymus DNA). The longer the substituent (methyl, ethyl, and propyl), the longer was the distance between charges of DNA and quaternized PVP because of the shielding, and the less was the complexation efficiency. Also, a decrease in charge density (PEVP-β, quaternization degree β = 23 or 46%) led to a decrease in PEC stability and a decrease in the critical salt concentration (salt concentration at which half of the EtBr molecules are intercalated in DNA-free sites).

Strong polycations containing aromatics or having a charged backbone: (a–c) Vinyl polymers containing aromatics and other architectures such as (d) ionenes, and (e) poly (N,N′-dimethyldiallylammonium chloride)
Given that poly(1-vinylimidazole) (PVIm) has a pK a of around 5.5, this polymer does not complex DNA at physiological pH. Thus, Allen et al. quaternized the imidazole ring with various substituents such as bromoethanol in order to obtain permanently charged imidazolium-containing copolymers [107]. As the quaternization degree was increased, fewer sites were available for protonation, thus the buffering capacity of the polymer decreased. With an increase in the quaternization percentage of PHEVIm-β, (Fig. 3b) the N:P ratio necessary for complexation with DNA decreased as well as the polyplex size (accompanied by a slight increase in zeta potential), suggesting a tighter binding between the polymer and pDNA. These effects reached a plateau at around 50% quaternization. At the same time, by increasing the quaternization percentage, the cytotoxicity increased (typical case). The maximum gene expression was observed for 25% quaternization for PHEVIm, which can be attributed to the right balance between PEC stability and efficient DNA release. To study the effects of adjacent hydroxyl number on transfection efficiency, two additional 25% quaternized copolymers, PEVIm-25, which did not contain hydroxyl groups, and PDHVIm-25 containing two hydroxyl groups for every four repeat units (n = 2) were compared to PHEVIm-25, which contained on average one hydroxyl group for every four repeat units (n = 1). As the number of hydroxyl groups increased, the initial N:P ratio required for polyplex formation decreased, suggesting hydrogen bond formation between the polycation and pDNA (Scheme 15). Indeed, previously, Reineke and colleagues found that the incorporation of hydroxyl groups further enhanced the binding of polymer to pDNA through hydrogen bonding and concluded that hydrogen bond formation between polymers and pDNA would serve as a less toxic alternative to high charge density polyelectrolytes [108].

(a) Structure–property–transfection relationships for imidazolium copolymers with controlled charge density and side chain hydroxyl number. (b) Cationic polymers electrostatically bind and condense anionic pDNA, forming a polyplex. Various factors, including hydrogen bonding, impact polyplex stability. Reprinted with permission from [107]. Copyright 2011 American Chemical Society
Regarding transfection efficiency, PEVIm-25 was two orders of magnitude less efficient than SuperFect tranfection reagent, while PHEVIm-25 was less than one order of magnitude less efficient and two orders more efficient than naked DNA in COS-7 cells; however, PDHVIm-25 was slightly less efficient than PHEVIm-25. Therefore, a balance has to be found between the hydrogen bonding properties of the polycation (facilitating DNA binding but not its release) and the shielding of the positive charge by the presence of hydroxyl groups, which reduces the protein adsorption and cytotoxicity but also the transfection efficiency. For the PVIm copolymers, one hydroxyl group in the form of PHEVIm seemed to be the optimal choice. This approach using hydroxyl groups to benefit from the hydrogen bonding capacity and decrease in toxicity has also been used with weak polyelectrolytes based on polymethacrylates [123, 124].
Ammonium- and phosphonium-containing polyelectrolytes (PTEA, PTBA, PTEP, and PTBP) differing in the nature of the quaternized group (ammonium versus phosphonium) and the length of their substituents (triethyl versus tributyl) were studied by the group of Long (Fig. 3c) [125]. According to gel electrophoresis, the ammonium polyelectrolytes bound DNA at higher +/− ratio compared to the phosphonium polyelectrolytes, which suggested improved DNA binding of phosphonium cations over ammonium cations. The authors proposed that a combination of different charge densities and cation sizes (phosphonium is a larger cation with less diffuse positive charge than ammonium) were responsible for the better DNA binding affinity of the phosphonium polyelectrolytes compared to ammonium. All polyelectrolytes condensed DNA into polyplexes of about 200 nm or less at +/− ratio of 4, except for PTBP, which condensed DNA into polyplexes of this size at +/− ratio of 6, meaning that the binding is less tight. PTEA, PTBA, and PTEP also exhibited a plateau in their zeta potential (positive) without significant change from a +/− ratio of 2. PTBP polyplexes generated at +/− ratio of 2 had zeta potentials near neutral, and then a positive plateau starting at +/− ratio of 4. The presence of a plateau in the polyplex diameter and zeta potential suggests that the additional polymer remained as free polymer in the solution, uncomplexed to DNA. The zeta potentials of the triethyl-based polyplexes were more positive than those of the tributyl-based polyplexes due to hydrophobic screening of the cationic charge with longer alkyl chains. All polymers exhibited similar toxicities due to their 100% charge densities, approximately like that of the transfection reagent jetPEI. PTEA and PTEP displayed poor transfection efficiency compared with SuperFect, whereas PTBA and PTBP exhibited excellent transfection, similar to SuperFect. Given that the entry into the cell of all polyplexes was successful, the higher transfection efficiency of tributyl-containing polyelectrolytes over triethyl-based polyelectrolytes could be due to a higher endosomolytic activity.
Ionene are polycations with charged quaternized nitrogen atoms in the polymer backbone (Fig. 3d). Izumrudov and colleagues synthesized ionenes via Menshutkin polyaddition reaction between N,N,N′,N′-tetramethylethylenediamine (TMED) and dibromoalkanes such as 1,4-dibromobutane and 1,8-dibromooctane [126]. For [ionene]/[DNA] < 1, the increase in ionene content was accompanied by a substantial decrease in PEC particle size (from 500 to 100 nm), up to a charge ratio of unity, where the particles were neutral and formed aggregates. With excess polycation, the positively charged PEC did not aggregate, and at charge ratios of the polymers of 2:1 the particle size was again ~100 nm, regardless of the charge density or chain length of the polycation. Nevertheless, a difference could be observed in the protection of DNA against nuclease attack: the polymers with the highest DP offered better protection and, at a given DP, the shortest spacer (i.e., the highest charge density) was preferred. These results correlated with the stability of the polyplexes, even if upon lengthening of the ionene chains (DP > 20), the difference in PEC stability between ionenes with different spacers became relatively small. The transfection efficiency in COS-7 cells followed the same trend for the ionenes as the PEC stability.
By comparing the DNA/polycation complexes based on various architectures such as PEVP (Fig. 3a), ionene (Fig. 3d) and PDMDAAC, which is a polycation of pendant type (Fig. 3e), Galaev and colleagues suggested that phase separation in solutions of DNA-containing PECs (with strong polycations without steric stabilization) follows the general rules ascertained from PECs formed by flexible vinyl polyanions. However, the high rigidity of the double helix of native DNA appears to be responsible for significant extension of the region of insoluble PECs at the expense of the region in which soluble PECs are formed [127].
2.1.1.2 With Side Chains Containing Quaternary Ammoniums
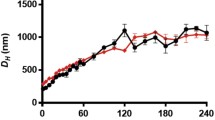
In regular DNA/polycation systems, at a charge ratio close to unity, macroscopic phase separation occurs: the cationic units of the polymers will form ion pairs with the anionic phosphate groups of DNA to yield charge-neutralized complexes. By the introduction of a sufficient amount of steric stabilizer such as PEG in the polycation (as block copolymer or grafts), this macroscopic phase separation can be avoided due to the lyophilizing effect of the PEG segments and the complexes will remain stable in solution. This was observed for P(TMAEMA-co-OEMA), M w = 2.8 × 105 g mol−1, 15 mol% oligo(ethylene glycol) grafts (OE), and 4–5 oligo(ethylene glycol)methyl ether units per graft (Fig. 4a) [128] compared to the non-PEGylated polymer at a ratio close to unity and for similar polymers, P(MAPTAC-co-OEMA), with higher degree of OE substitution, 8 oligo(ethylene glycol)methyl ether units per graft, M w = 1.8 × 106 g mol−1 for 89 mol% OE, and M w = 3.5 × 106 g mol−1 for 94 mol% OE (Fig. 4b) [129]. Complete binding of DNA by these polycations occurred at a ratio of more than 1 due to the presence of PEG, which partially screens the charges of the polycation (the higher the PEG content, the higher the ratio for complete binding). At a charge ratio of ~2 for these P(MAPTAC-co-OEMA) copolymers, the zeta potential reached a plateau at a neutral value and the hydrodynamic diameter stayed constant, meaning that the excess polycation was not incorporated into the polyplex. On the contrary, for the examples of the last paragraph that did not possess a steric component and for which the same phenomenon had mainly an electrostatic explanation, the lack of incorporation of the excess polycation into the polyplex occurred in this case mainly because of the steric repulsion produced by the PEG corona. Moreover, the DNA present in these polyplexes was inaccessible to DNAse I, which clearly indicates that the PEG segments present in the outer part of the polyplexes protect the DNA inside the polyplexes. Nevertheless, it is surprising that despite the high content of PEG in the polycation, the DNA binding was still efficient. By contrast, for P(MAPTAC-co-OEMA) the zeta potential was positive at a high charge ratio of random copolymers, P(TMAEMA-co-HPMA) (Fig. 4c) [130]. Nevertheless, only binding of DNA is not enough and a tight binding is desired. The copolymers P(TMAEMA-co-HPMA) containing the lowest degree of ammonium (5 and 15%) showed virtually no ability to displace EtBr and also did not protect DNA from degradation by endonucleases, probably because their association with DNA was too weak. The inverse tendency has been observed for PLL and PLS and their trimethylated derivatives (PtmL and PtmLS, respectively; Fig. 4d) [131]: the complexes with trimethylated peptides seemed to be looser (according to EtBr complexation and exchange reaction with anions) but the compaction of DNA occured at lower charge C/A ratio, due to their higher charge density.Interestingly, the transfection efficiency of the trimethylated PtmLS into COS-1 cells was better than that of PtmL and the non-quaternized derivatives, despite their similar intracellular distribution. Thus, it seems that a loose structure for the release of DNA from the complex (at best from endosome into the cytoplasm) is necessary, as well as the presence of functional groups such as serine residues that impart hydrophilicity and hydrogen bonding capacity.

(a–f) Strong polycations with side chains containing quaternary ammoniums: Methyl methacrylate and amide backbones
A similar construct to PLL, based on a slightly different amide backbone, a polyaspartamide derivative containing a quaternary ammonium, α,β-poly{(N-2-hydroxyethyl)-N-carbazate[N-(3-trimethylammonium chloride) propylhydrazide]-d,l-aspartamide} (PHEA-HYD-CPTA) (Fig. 4e) [132], also revealed itself to efficiently complex DNA and reduce its rate of degradation by DNAse. Similar polymers but with a block structure were also efficient at protecting DNA against enzymatic degradation. Indeed, partially methylated PEG-mPDMAEMA-β and completely methylated PEG-mPDMAEMA or butylated PEG-bPDMAEMA with PEG blocks of various lengths (Fig. 4f) [133] formed, as previously explained, micellar-type structures with a PEG protective shell that is more difficult to achieve for polymers grafted with oligoethylene glycol segments.
2.1.2 Weak Polyelectrolytes
2.1.2.1 Without Steric Stabilizer
2.1.2.1.1 Polyaspartamide Derivatives
PAsp-DETA (Fig. 5a), a polyaspartamide derivative [134] that is degradable and thus causes minimal toxicity, shows a higher transfection efficiency in HUVEC cells after repeated administration compared to linear PEI (22 kDa) and PGlu-DETA (Fig. 5b) that are non-biodegradable. Among polyaspartamides modified with oligoethyleneimine side chains of various lengths [ethylene diamine (PAsp-EDA) in Fig. 5c, and triethylene pentamine (PAsp-TEPA), pentaethylene hexamine (PAsp-PEHA), and polyethyleneimine (PAsp-PEI) in Fig. 5d] [135], PAsp-PEHA showed the highest capacity of condensation, similar to PAsp-PEI (diameter <300 nm), while PAsp-EDA showed the lowest DNA condensation capacity. Full retardation of DNA migration occurred at N:P = 1:1 for PAsp-TEPA, PAsp-PEHA, and PAsp-PEI, and only at 5:1 for PAsp-EDA. This trend was also observed for the transfection efficiency in HEK293 cells, meaning that the length of oligoethyleneimine side chains is an important factor and that a side chain with four ethylene imine repeating units was enough for condensation. Similar polyaspartamide derivatives (α,β-poly[(N-2-hydroxyethyl)-d,l-aspartamide]) but with spermine side chains (and not oligoethylene imine) were studied by the group of Cavallaro, i.e., α,β-poly[(N-2-hydroxyethyl)-d,l-aspartamide] modified with spermine (PHEA-Spm) (Fig. 5e) and α,β-poly[(N-2-hydroxyethyl)-d,l-aspartamide] modified with spermine-butyramide (PHEA-Spm-C4) (Fig. 5f) [136]. In this case, full retardation of DNA migration was observed at C:P = 0.75 for PHEA-Spm and 2 for Spm-C4. The zeta-potential values became positive at a polycation:DNA weight ratio above 1.5 for PHEA-Spm, and 2.5 for PHEA-Spm-C4. These values are in agreement with the lower amount of free primary amino groups present in PHEA-Spm-C4 compared with PHEA-Spm. A nearly total quenching of EtBr was reached at C:P = 8 for PHEA-Spm, and at 10 for Spm-C4, but at the same time the condensing ability of PHEA-Spm-C4 was superior to that of PHEA-Spm; at C:P = 1 the polyplex diameter was 600 nm for PHEA-Spm and 130 nm for PHEA-Spm-C4. It seems that the introduction of a short hydrophobic chain in the structure enabled an increased condensing capacity, probably conferred by hydrophobic interactions with DNA, which in turn led to a decrease in transfection efficiency, which could be due to the stability of the polyplex (even if EtBr displacement shows equivalent performances).

(a–f) Weak polycations without steric stabilizer: Polyaspartamide and polyglutamide and their derivatives
2.1.2.1.2 Other Polyamide Backbones
Poly(amidoamine)s with pendant primary amines [amino ethane (PA-AE), amino butane (PA-AB), and amino hexane (PA-AH)] (Fig. 6a) [137] all had a better buffering capacity than branched PEI of 25 kDa. Those with the longest alkyl chains had higher buffering capacity than PA-AE in the pH range 5–7, which is important for endosomal release. Cytotoxicity in 293T and COS-7 cells was concentration dependent and proportional to the length of the alkyl chain: the longer the chain, the more toxic. As in the case of polyaspartamide modified with oligoethyleneimine side chains of various lengths [135], the shortest chain showed the lowest condensation capacity (even if no secondary amines were present in the side chains of these poly(amidoamine)s with pendant primary amines). This suggests that the condensation capacity (size of the polyplex) was mainly a function of the accessibility and degree of protonation of the primary and tertiary amines. Polymers with longer chains showed better performances until a certain chain length, given that when the flexibility of the chains was sufficient, no further improvements were observed (zeta potentials had the same profile). These results showed that these poly(amidoamine)s with pendant primary amines were a bit less efficient than polyaspartamide derivatives of comparable length regarding full retardation of DNA migration. Indeed, DNA was fully retarded at N:P = 5 for PA-AE, 3 for PA-AB, and 2 for PA-AH; this could be explained by the absence of secondary amines in the side chains. Despite the better performances of PA-AH, its transfection efficiency in 293T cells was lower than that of PA-AB, probably due to its higher cytotoxicity or its slightly lower buffering capacity. It is important to note that the transfection efficiency of these polymers was dependent on the cell type (low transfection efficiency in COS-7 cells compared to 293T).

(a–c) Weak polycations without steric stabilizer: Polyamide backbones
Comparable bioreducible [138] poly(amidoamine)s with oligoamine side chains (ethyleneimine or spermine type) were studied by Engbersen and coworkers [139]. The introduction of disulfide linkages in the backbone is a way to reduce the cytotoxicity by promoting its biodegradability in the reducing intracellular environment, in contrast to the last example [137]. Moreover, the cleavage of the backbone is supposed to promote DNA release intracellularly, thus improving the transfection. Indeed, poly(amidoamine)s with pendant ethylene diamine (SS-PA-EDA), diethylene triamine (SS-PA-DETA), triethylene tetramine (SS-PA-TETA), and tetraethylene pentamine (SS-PA-TEPA) (Fig. 6b) showed extremely low cytotoxicity in COS-7 cells even at 50 μg/mL (more than 90% cell viability reported, but duration of exposure was not clear) [139]. Nevertheless, poly(amidoamine)s with pendant norspermidine (SS-PA-NSpm) or spermine (SS-PA-Spm) (Fig. 6c) showed much higher toxicities. The enlargement of the alkyl spacer between the amino groups in the side chain from ethylene to propylene seems to be responsible for the higher cytotoxicity. When one compares the cytotoxicity of SS-PA-EDA, SS-PA-DETA, SS-PA-TETA, and SS-PA-TEPA in COS-7 cells to PA-AE, PA-AB, and PA-AH [137], the two last polymers showed much higher toxicities at the same concentration after 24 h exposure, which might only be an effect of duration of exposure and a time-dependent cytotoxicity. If not the duration, this could be a positive effect of the disulfide bond or the lower M w. Except for SS-PA-EDA, these poly(amidoamine)s showed comparable or slightly higher buffering capacities than branched PEI of 25 kDa. Similar results as for poly(amidoamine)s with pendant primary amines concerning DNA retardation in gel electrophoresis were obtained with the best polymer, SS-PA-TEPA (N:P = 2). All these polycations condensed pDNA in a similar manner as regards the diameter of the polyplexes (<150 nm). The polyplexes of the SS-PA bearing secondary amines in their side chain (SS-PA-DETA, SS-PA-TETA, and SS-PA-TEPA) induced relatively high transfections, but those of SS-PA-EDA with only a terminal primary amino group in the side chain gave only low transfection efficiency, reinforcing the idea that a minimum chain length is needed and/or the presence of secondary amines and higher buffering capacities of the polymers, suggesting a more facilitated endosomal escape of their polyplexes. Enlargement of the alkyl spacer between the amino groups in the side chain from ethylene to propylene (SS-PA-NSpm and SS-PA-Spm) had a negative effect on transfection efficiency. The reason was unclear given that polyplex size, surface charge, and buffering capacity did not deviate significantly from the other SS-PA. This could be due to their inherent cytotoxicity.
2.1.2.1.3 PDMAEMA and Derivatives
Poly[2-(dimethylamino)ethyl methacrylate] (PDMAEMA) is usually used as weak polyelectrolyte for gene delivery and has the additional property of being temperature sensitive [140, 141]. The study by Stolnik and coworkers on PDMAEMA (Fig. 7a) emphasized the condensation behavior of PDMAEMA as a function of pH [102]. As can be intuitively understood, the ionization of PDMAEMA increases from pH 8 (only 24% ionization) to pH 4 (polycation nearly completely ionized), therefore the binding of PDMAEMA with DNA is tighter (EtBr displacement assay) at lower pH, which is “counterproductive” when it comes to release of the genetic material. But, this effect is balanced by its buffering capacity via the tertiary amine groups, which is favorable for endosomal escape.

(a–m) Weak polycations without steric stabilizer (except (g) and (j) for comparison): PDMAEMA and derivatives
PDMAEMA of various molecular weights were tested by the group of Hennink for their transfection efficiency [142]. In COS-7 and OVCAR-3 cells, high molecular weight polymers (M w < 300 kDa) were more efficient in transfection than the low molecular weight polymers (M w < 60 kDa), which was related to their property as condensing agents. Low molecular weight polymers led to polyplexes with sizes bigger than 300 nm (up to 1 μm), while high molecular weight polymers gave polyplexes with sizes in the range 150–200 nm, and these smaller particles seemed to enter the cells more easily.
PDMAEMA and its derivatives PDMAPMAm, PDMAPMA, PDMAEMAm, and PTMAEMA of higher molecular weight (M n > 25 kDa) than in the first study or of comparable molecular weight in the case of PDMAEA and PDEAEMA (M n < 10 kDa) (Fig. 7b–e) were studied by Hennink and colleagues [143]. The methacrylate/methacrylamide derivatives of PDMAEMA of high molecular weight were able to condense pDNA, yielding polyplexes with sizes of 100–300 nm and a slightly positive zeta potential, while PDMAEA and PDEAEMA were not capable of condensing pDNA to small particles, possibly due to their relatively low molecular weight (and low solubility of PDEAEMA at pH 7). The transfection efficiency and the cytotoxicity of the polymers differed widely: the highest transfection efficiency and cytotoxicity were observed for PDMAEMA. As PDMAEMA is capable of condensing DNA to small particles and has the lowest average pK a value (7.5) of all condensing derivatives, PDMAEMA has the highest buffering capacity and probably behaves as the best candidate for endosomal escape. Furthermore, molecular modeling showed that, of all studied polymers, PDMAEMA has the lowest number of interactions with DNA. Therefore, the authors hypothesized that the superior transfection efficiency of its polyplexes can be ascribed to the intrinsic property of this polymer to destabilize endosomes combined with an easy dissociation of the polyplex once present in the cytosol and/or nucleus.
Hennink and colleagues also studied copolymers of DMAEMA with various monomers such as the hydrophobic methyl methacrylate (MMA) in P(DMAEMA-co-MMA) (Fig. 7f), or hydrophilic N-vinyl pyrrolidone (NVP) in P(DMAEMA-co-NVP) (Fig. 7h), and OEGMA in P(DMAEMA-co-OEMA) (Fig. 7g) of M w > 90 kDa [142]. A copolymer with 20 mol% of MMA showed reduced transfection efficiency and a substantially increased cytotoxicity compared with PDMAEMA of the same molecular weight. A copolymer with OEGMA (48 mol%) showed both a reduced transfection efficiency and a reduced cytotoxicity (presence of OEMA), whereas a copolymer with NVP (54 mol%) yielded smaller particles at a lower P:DNA ratio than PDMAEMA or than the other copolymers with equivalent degree of modification (as NVP interacts with DNA via hydrogen bonding) and showed an increased transfection and decreased cytotoxicity.
Further derivatives of PDMAEMA were synthesized in order to improve its condensation ability at physiological pH, such as a comb-type polycation consisting of P(DMAEMA-co-PLL) (Fig. 7i) [144]. This copolymer possessed a pH-dependent behavior due to the presence of PDMAEMA (pK a 7.5) and PLL (pK a 10). The presence of the PLL segments prevented precipitation of the copolymer at pH > 7.5, as is the case for PDMAEMA homopolymer, and this comb-type polymer was capable of DNA condensation at pH 8 (as observed using circular dichroism).
Poly(hydroxyethyl methacrylate) backbones, onto which poly[2-(dimethylamino)ethyl methacrylate] of various lengths were grafted via click chemistry [P(HEMA-g-PDMAEMA), M w > 75 kDa; Fig. 7j], were able to condense DNA into small particles (90–190 nm, at a polymer to plasmid mass ratio of 6) [145] and the sizes as well as the zeta potentials of the P(HEMA-g-PDMAEMA)-based polyplexes were independent of the molecular weight of P(HEMA-g-PDMAEMA) (for M w of 75 kDa and above). P(HEMA-g-PDMAEMA) showed more EtBr quenching than the starting PDMAEMA, indicating a weaker binding of this low molecular weight polymer to the pDNA. However, it showed similar quenching as high molecular weight PDMAEMA, but less toxicity.
A study comparing PDMAEMA with PEGylated derivatives and different functional groups such as tertiary amine, pyridine, imidazole, and acid groups was conducted by Schacht and colleagues [146]. They found that the presence of methacrylic acid in P(DMAEMA-co-MA) (Fig. 7k) increased the amount of polymer needed for DNA condensation with increasing amount of acid groups, which can be explained by the repulsive effect between anionic DNA and the negatively charged carboxylate groups. A similar effect was observed for imidazole-containing polymers, as the imidazole groups are not protonated under the experimental conditions. These results also correlated with the zeta potential measurements: the greater the amount of these groups (negatively charged or neutral), the lower the zeta potential of the polyplexes. P(DMAEMA-co-MA), P(DMAEMA-co-HYMIMMA) (Fig. 7l), and P(DMAEMA-co-HENIMA) (Fig. 7m) were able to condense DNA into nanoparticles with size <300 nm at a charge ratio of 2:1 but with bimodal or trimodal distributions (not well-defined, possibly due to aggregation).
2.1.2.1.4 PNIPAM Derivatives
Homopolymers of PNIPAM have a lower critical solution temperature (LCST) around body temperature. The LCST of copolymers of PNIPAM such as P(DMAEMA-co-NIPAM (Fig. 8a) gradually increased with increasing content of DMAEMA (hydrophilic monomer) and was independent of the molecular weight (38.3–45.7°C for a DMAEMA content of 15–30% and >80°C for a content of 80%) [147]. All P(DMAEMA-co-NIPAM) copolymers (M w > 91 kDa), even with a low DMAEMA content of 15 mol%, were able to bind to DNA. With increasing NIPAM content, the charge density of the copolymer decreased and the P:DNA ratio needed for condensation to occur increased. Concerning the polyplexes, with increasing NIPAM content, the zeta potential of the polyplexes at the optimum P:DNA ratio decreased, as did the cytotoxicity and transfection efficiency. The authors postulated that these polyplexes interacted less with the negatively charged membrane, thus leading to lower transfection efficiency. At 37°C, even if the LCST was not passed, complexes made from low molecular weight polymers (independent of the content) or of high molecular weight with low DMAEMA content (15%) showed poor properties as transfection agents, which was linked to their poor stability.

(a–c) Weak polycations without steric stabilizer: PNIPAM derivatives
The condensation properties of PLL-g-PNIPAM (Fig. 8b) were governed, like in the last case, by the PNIPAM content related to the condensing units such as lysine (higher content of PNIPAM, less condensation efficiency) but the molecular weight of the PNIPAM grafts did not have a significant effect [148]. As also observed in the previous study [147], the size of the complexes increased with increasing PNIPAM content at 25°C due to an internal swelling of the hydrated PNIPAM chains. At 38°C (above the phase transition of PNIPAM), the size of the polyplexes decreased as a consequence of the collapse of PNIPAM chains, accompanied by a higher structural density and thus a more difficult release of DNA.
Similar architectures based on polyarginine of shorter length than PLL, P(LArg)-g-PNIPAM (Fig. 8c), showed comparable results concerning the physico-chemical characteristics of the polyplexes and were influenced by the LCST of PNIPAM (LCST of P(LArg)-g-PNIPAM was 35.2°C) [149]. The low cytotoxicity of P(LArg)-g-PNIPAM was also attributed to the reduction in the positive charge density of polyarginine upon incorporation of neutral PNIPAM chains. Transfection studies in COS-1 cells showed that temporary cooling below the LCST of P(LArg)-g-PNIPAM was favorable for gene expression and that via this method, at an appropriate complexation ratio, the transfection efficiency of P(LArg)-g-PNIPAM was equivalent to that of Lipofectamine 2000.
2.1.2.1.5 PLL Derivatives
Derivatives of PLL containing endosomal escape moieties such as imidazole or histidine are presented in this section. Copolymers of PLL-g-Im (Fig. 9a) with various content of imidazole (>73.5%) were all capable of condensing DNA but at higher ratios and bigger size of polyplexes than PLL alone [150]. Nevertheless, they all had a very low cytotoxicity in various cell lines (CRL1476 smooth muscle cells, P388D1 macrophages, HepG2 hepatoblastoma) compared to PLL, which cannot be explained by big discrepancies in the zeta potentials. At pH 7.2, approximately 5% of the imidazole groups on the polymer are protonated and are potentially available to assist the PLL moieties in the condensation of DNA. Interestingly, with a 9% increase in imidazole content (from 73.5 to 82.5 mol%), the level of luciferase expression approximately doubles at each DNA:polymer ratio. However, with a further 4% increase in imidazole content (from 82.5 to 86.5 mol%), luciferase expression levels approximately double again. These results suggest a nonlinear relationship between polymer imidazole content and protein expression. Later studies of PLL-g-Im with other M w and percentage of grafting (Fig. 9a) gave more insights into this phenomenon [151]. The studies showed that the polymer cytotoxicity was reduced when the polymer was complexed with pDNA and decreased with increasing imidazole content and decreasing molecular weight. As in the previous study [150], the polymers with the highest IC50 (i.e., lowest toxicity and highest imidazole content) also mediated the highest level of protein expression in NIH-3T3 cells, probably due to the endosomal escape capacity of imidazole groups. But, increased protein expression in a culture population did not always correlate with an increased number of transfected cells. As the imidazole content increases, the polymer becomes less cationic and is therefore less able to electrostatically interact with DNA and quench EtBr fluorescence (condensation); at the same time, the relative binding affinity for DNA increases with increasing imidazole content for polymers of various molecular weights, which suggests that other intermolecular forces play a role [152].

(a, b) Weak polycations without steric stabilizer: PLL derivatives
Similar results concerning the cytotoxicity were found for PLL-g-PHis (Fig. 9b) [153], which are in line with the transfection efficiency. Once more, the decrease in cationic charges due to the substitution of PLL backbone led to a lowering of the cytotoxicity, and the introduction of histidine (imidazole ring) led to better transfection efficiency due to its buffering capacity, i.e., endosomal escape properties.
2.1.2.1.6 Other Amino Acid-Based Polymers
Generally, polymers modified with histidine or other moieties containing an imidazole group have shown significant enhancement of gene expression without increased cytotoxicity, compared with non-modified polymers, but can present solubility problems [154–160].
Schacht and colleagues. studied (co)polymers of dimethylaminoethyl-l-glutamine (PDMAEG, Fig. 10a) and l-glutamic acid [P(DMAEG-co-Glu); Fig. 10c-R 2], partially grafted with various moieties such as pyridine in P(DMAEG89%-co-PyEG11%) (Fig. 10c-R 1) [161], primary amine in P(DMAEG85%-co-AEG15% (Fig. 10e-R 1), imidazole in P(DMAEG-co-HisG) (Fig. 10e-R 2), and ethylguanidine in P(DMAEG83%-co-AgmG17%) (Fig. 10e-R 3) [162] and compared them with PDMAEG and P(DEAEG67%-co-LLys33%) (Fig. 10b) [161] as well as with PHisG (Fig. 10d) and poly[(histamino-l-glutamine)73%-co-(agmatino-l-glutamine)27%] (P(HisG-co-AgmG); Fig. 10f) [162]. The mass per charge for all the copolymers was in the range 222–290 Da. The highest condensations (EtBr) were obtained for PDMAEG and P(DEAEG-co-LLys) compared to P(DMAEG-co-PyEG) and P(DMAEG-co-Glu) (but all gave <50% EtBr fluorescence), respectively, because of the highest percentage of tertiary amines among the PDMAEG derivatives and the presence of the primary amines of PLL [161]. All the copolymers were degradable under biologically relevant conditions in a time frame varying from hours to days, and formed polyplexes with DNA with a diameter <100 nm. The most potent polymers in transfection studies in HEK293 cells were the polymers containing pyridine and acid groups (by a factor of ten) and not PDMAEG or P(DEAEG-co-LLys) [162]. The explanation for the better performances of the pyridine derivatives was not clear, given that the pyridine moiety did not provide extra buffering capacity. The potency of the polymers containing a carboxylic acid group could be because it eventually destabilized the cell membranes in its carboxylate form.

(a–j) Weak polycations without steric stabilizer: other amino acid-based polymers such as polyglutamic acid, polyornithine, polyhistidine, etc.
Polymers containing more than 70% imidazole were not able to condense DNA (>50% EtBr fluorescence), i.e., PHisG, P(HisG73%-co-Agm27%), or the copolymers from the P(DMAEG-co-HisG) series [162]. PDMAEG, P(DMAEG85%-co-AEG15%), P(DMAEG82%-co-HisG18%), and P(DMAEG64%-co-HisG36%) all possessed a similar charge ratio, causing 50% reduction of EtBr fluorescence (+/− around 0.8), and this value increased with increasing imidazole content; P(DMAEG84%-co-AgmG16%) showed a higher value than P(DMAEG85%-co-AEG15%). According to the authors, this might be due to the longer distance between the charged guanidine group and the main polymer chain in comparison with the polymers containing tertiary and primary amines, resulting in a weaker electrostatic interaction between the polymer and the DNA. On the same line, the smallest complexes were formed with P(DMAEG85%-co-AEG15%), probably because of the primary amines that allow a better interaction with the DNA in comparison with the tertiary amines (less steric hindrance, higher protonation degree). PHisG and P(His73%-co-AgmG27%) formed the largest complexes. In the case of pHisG, the large size could be due to the weak interaction between the polycation and the DNA (few imidazole functions are protonated). The large complexes formed with P(His73%-co-AgmG27%) could be explained by the fact that the zeta potential of the complexes was close to neutrality, leading to aggregation of the complexes. All the polyplexes, except those based on P(HisG73%-co-AgmG27%), had low transfection efficiency in COS-1 cells, which could be due to their poor ability to interact with the membrane of the cells and thus the polyplexes were not taken up by the cells [163]. On the other hand, polyplexes based on PHisG73%-co-PAgmG27% (more cytotoxic) at a ratio of 8:1 were more efficient than PEI-DNA at a ratio of 2:1, which may be due to the presence of the guanidine functions (more pronounced positive charge of the guanidine functions, pK a = 12.5, in comparison with tertiary amines, pK a = 10).
Other imidazole derivatives based on poly(l-histidine) partially grafted with gluconic acid (PHis-g-Glu, Fig. 10i) to impart solubility at physiological pH (polyhistidine is insoluble in aqueous solutions at pH >6.0) were reported by Pack et al. [164]. Approximately one carbohydrate–imidazole substitution out of every 23 imidazole groups was enough to confer solubility of at least 100 mg mL−1 at pH 7. Migration of DNA was completely retarded at a pHis-g-Glu:DNA weight ratio of 3:1, which was consistent with the charge ratio causing 50% reduction of EtBr fluorescence; the size of the polyplexes at this ratio was >500 nm. The DNA seemed to be fully condensed at a pHis-g-Glu:DNA weight ratio of 5:1, given that the size of the polyplexes was 240 nm at pH 7. Unfortunately, these polyplexes were unable to transfect COS-7 cells, maybe because of their large size, despite their negligible cytotoxicity.
Poly(l-ornithine) (PLO, Fig. 10g) has a structure similar to polylysine, except that it possesses one CH2 less in the side chain. In a publication by the group of Gumbleton, polyplexes based on PLO, PLL and poly(d-lysine) (PDL) possessed roughly similar physico-chemical characteristics; nevertheless, the polyplexes based on PLO showed better transfection capacity in A459cells and COS-7 cells [165]. It should be noted that condensation of pDNA occurred at the following charge ratios (<10% EtBr fluorescence): 0.8:1 for PLO, 1.2:1 for PLL, and 1.5:1 for PDL, and that PLO polyplexes also showed greater resistance to polyanion-induced disruption. As explained by the authors, Blauer and Alfassi had previously suggested that the additional CH2 group contained within the lysine may make the α-helix conformation in PLL more stable than in PLO [166]. Given the comparable pK a values of the primary amine groups for lysine and ornithine (pK a = 10.5–10.7) [167], it is probable that conformational differences rather than protonation per se provides a basis for the differential behavior of PLO-mediated pDNA condensation. The least effective condensing polycation was PDL. Given that the l-isomer is the natural orientation of nuclear enzymes and proteins, it is possible that DNA interactions with other macromolecules are biased towards l-isomer conformations [168].
In order to determine the size of the polyplexes, most of the studies use light scattering, which gives the R h as well as other information, but is not able to give the exact structure of the polyplexes. Therefore, AFM is a complementary method to light scattering and allows imaging matter at the nanoscale (possible interactions with the surface of the wafers should nevertheless be taken into account). An AFM study of the complexation of DNA by PLO was conducted by the group of Ganguli and could provide insights into the mechanism of DNA condensation [169]. Based on AFM images, the mechanism seemed to be different at low (<7 μg mL−1) and high DNA concentrations (>13 μg mL−1), i.e., monomolecular and multimolecular condensations, respectively (Scheme 16). It should nevertheless be noted that in contrast to most of the studies where the polycation is added to DNA solution, in this study DNA was added to the polymer solution, which could influence the mechanism pathway.

Proposed mechanism of condensation of plasmid DNA with poly-l-ornithine at high (13–20 μg mL−1) and low DNA (3–7 μg mL−1) concentrations. Monomolecular condensation is seen at low DNA concentration whereas both monomolecular and multimolecular condensation are seen at high DNA concentrations. Reprinted with permission from [169]. Copyright 2007 Elsevier
A low molecular weight PAGA (Fig. 10h) has also been tested [170]. The structure of PAGA is similar to PLL except for the backbone linkages, which are ester bonds for PAGA and peptide bonds (amide) for PLL. PAGA strongly condensed DNA at a charge ratio of less than 2 (<10% EtBr fluorescence), with a charge ratio of around 1 causing 50% reduction in EtBr fluorescence (results in the same order as PLL). The fast degradation of PAGA compared to PLL was suggested to be a way to reduce toxicity and lead to a faster release of DNA from the polyplexes after internalization by the cells, and was confirmed in 293 cells. The transfection efficiency of PAGA was indeed three times higher than PLL under optimized conditions (PLL, charge ratio 6; PAGA, charge ratio 60). According to the authors, the tenfold charge ratio required for PAGA compared to that for PLL was the result of the premature degradation of PAGA.
Poly(4-hydroxy-l-proline ester) (Fig. 10j) of low molecular weight, another polyester based on amino acids, was also hypothesized as a good candidate due to its degradability, and indeed achieved low cytotoxicity in COS-7 cells [171]. Complete DNA retardation occurred at weight ratio of 1:1 where the polyplexes were neutral and therefore aggregated (size >1,300 nm) but efficient condensation was observed at polymer:DNA ratio of 2:1 (size of the polyplex <200 nm). Unfortunately the transfection efficiency of these polyplexes was not reported.
Peptoids (poly-N-substituted glycines) are a class of peptidomimetics whose side chains are appended to the nitrogen atom of the peptide backbone, rather than to the α-carbons [172]. Despite their relatively tedious synthesis, there are prospective applications in the biomedical field, for instance in gene therapy [173, 174]. The most efficient peptoid from a vast series as DNA condensing agent was based on the repetition of N-(2-aminoethyl)glycine (Nae) and N-(2-phenylethyl)glycine (Npe), (NpeNpeNae)12, showing the importance of the presence of primary amines and hydrophobic motifs [175]. The authors could show that the spacing of charged residues on the peptoid chain as well as the degree of hydrophobicity of the side chains had much influence on the ability to form homogeneous complexes with DNA. Moreover, the authors found that the transfection efficiency was highly dependent on the primary sequence of the peptoid and, to a lesser degree, on the length of the peptoid. Zuckermann and colleagues studied another series of peptoids based on the alternance of primary amine and hydrophobic groups, either phenyl or isopropyl, and lipitoids (peptoid–phospholipid conjugates) starting from these peptoids [176]. At charge ratios above unity, only (NpeNpeNae)12 and (NaeNpeNpe)12 and both lipitoids were efficient in inducing transfection, which was correlated with significant cytotoxicity. Unfortunately, it was not possible in this study to correlate the physical properties of peptoid/lipitoid:DNA complexes with their transfection capabilities.
2.1.2.1.7 PMMA and Methacrylamide Derivatives
For a series of PAEM homopolymers (Fig. 11a) with various molecular weights [177], the ability to condense DNA and resistance against heparin destabilization increased with increasing molecular weight (retardation of DNA in gel electrophoresis at a ratio N:P of 1:1 for PAEM75 and PAEM150, and at a ratio 2:1 for PAEM45). Regardless of PAEM chain length, the size of the polyplexes were <200 nm for a wide range of N:P ratio, their zeta potentials at N:P ratio of 8:1 were roughly similar, as was their cytotoxicity. On one hand, longer PAEM chains enhanced cellular uptake and nuclear localization of the polyplexes (probably linked to the greater stability of the polyplexes that might interact more strongly with membranes, according to the authors), while on the other hand shorter PAEM chains facilitated intracellular dissociation (more easily displaced from the polyplex). Nevertheless, even if the ideal carrier should posses both properties, the polymer with the longest chains also showed the highest transfection efficiencies in dendritic cells.

(a–g) Weak polycations without steric stabilizer: PMMA and methacrylamide derivatives
PHisA (Fig. 11a) is a water-soluble polymer possessing buffering capacity in the endosomal pH range [178]. All PHisA polymers with M n in the range of 9.2–28.7 kDa were able to completely inhibit DNA migration at a N:P ratio of 5:1 (higher ratios than for PAEM, containing primary amines [177]). At N:P ratios <15:1, the size of PHisA polyplexes decreased with increasing molecular weights from 9.2 to 17.5 kDa, whereas 17.5 and 28.7 kDa PHisA condensed DNA into comparable particle sizes at the same N:P ratios, as well as with increasing N:P ratio. For PAEM (M n = 9.8–33.7 kDa)-based polyplexes, the size was nearly independent of chain length or charge ratio. At an N:P ratio of 15:1 and above, all PHisA polymers were able to condense DNA into nanosized particles (<220 nm), and the sizes as well as zeta potentials reached a plateau (moderate positive surface charges of 13–19 mV). Although less toxic than PEI25K, by comparing the transfection efficiency of pHisA180 and pHisA300 in COS-7 cells to that of PEI25K at N:P ratio of 10, it was shown that their efficiency was slightly higher than for PEI, which could be attributed to their higher buffering capacity.
Preliminary results were reported on PHEMA substituted with amino acids such as glycine [P(HEMA-Gly), Fig. 11c-R 1], alanine [P(HEMA-Ala), Fig. 11c-R 2], valine [P(HEMA-Val), Fig. 11c-R 3], phenylalanine [P(HEMA-Phe), Fig. 11c-R 4], and lysine [P(HEMA-Lys, Fig. 11c-R 5] [179]. These polymers were able to condense DNA, while having very little toxicity (up to 250 μg mL−1 tested on COS-7 and SPCA-1 cell lines); unfortunately, only preliminary results were reported and there was no comparison between these amino acid-substituted PHEMAs, despite their interesting structures.
Several poly(methacrylate)- and poly(methacrylamide)-based homopolymers with various side chains bearing primary, tertiary, and quaternary ammonio groups were designed by the group of Seymour in order to study the influence of (1) length of side chains bearing cationic residues; (2) the nature of the amine, i.e., primary, secondary, or tertiary amines and quaternary ammonio groups; (3) charge spacing along the polymer backbone; and (4) molecular weight or degree of polymerization [180]. These polymers are presented Fig. 11d–g: PVA.HCl (Fig. 11d), PAA.HCl (Fig. 11e), PMAEDA.HCl (Fig. 11f-R 1), PMAGEDA.HCl (Fig. 11f-R 2), PMADGHDA.HCl (Fig. 11f-R 3), PDMAEMAm (Fig. 11f-R 4), PTMAEM.Cl (Fig. 11f-R 5), P(HPMA-co-TMAEM.Cl (Fig. 11g), and PBTMAIPM.I2 (Fig. 11f-R 6). At an N:P ratio of 2, considering first the influence of side-chain length, polymers with long side chains such as PMADGHDA.HCl were incapable of efficient complex formation with DNA (considerable residual EtBr fluorescence) but did show considerable transfection activity (positively charged complexes, thus their uptake by cells may be facilitated), despite their poor gene expression via direct intranuclear injection. At the other extreme, complexes formed using polymers with very short side chains, such as PVA.HCl and PAA.HCl, were efficient at complex formation (little residual EtBr fluorescence) and were remarkably stable to polyanion-mediated disruption. Efficient charge neutralization may explain their lack of surface charge, which probably underlies their tendency to aggregate. Their low transfection activity could be a consequence of their low surface charge (not uptaken by cells to a great extent), but their ability to undergo efficient intranuclear transcription was surprising in light of their stability to polyanions, as mentioned by the authors. The influence of cationic charge strength, using primary or tertiary amines or ammonio groups, on the properties of complexes formed with DNA was investigated. Polymers containing ammonio groups such as PTMAEM.Cl complexed DNA relatively efficiently, reflecting a stronger bond than with primary amino groups. The transfection activity of their complexes was 10- to 100-fold less than complexes based on most other cationic polymers. This poor transfection activity may be the result of poor access to the cytoplasm/nucleus, due to either cytotoxicity or poor endosomal release due to the absence of pH responsiveness (no proton sponge effect). By contrast, PDMAEMam containing tertiary amines was less effective at DNA condensation but showed good transfection activity in HEK 293 cells in vitro, probably because of the pK a of its amines in the endosomal range. In the next step, the influence of the density of positive charge along the cationic polymer on the properties of complexes formed with DNA was investigated. Comparing PTMAEM.Cl with IBTMAIPM.I2, which bears two ammonio groups per monomer instead of one, it was found that the properties of the polyplexes were remarkably similar, suggesting that after reaching a certain density of positive charges, the effects (DNA condensation, cytotoxicity, etc.) might plateau. The effect of charge dilution was addressed using random copolymers containing TMAEM.Cl and HPMA as monomers. All these copolymers (TMAEM.Cl, 5–75%) were capable of binding DNA, although copolymers with greater than 50% TMAEM.Cl content were not capable of forming particulate complexes (as shown by AFM). According to the authors, it seems likely that these copolymers could not drive hydrophobic self-assembly of polymer/DNA complexes (because of the presence of hydrophilic HPMA). Moreover, the complexes based on these random copolymers showed low levels of transfection similar to the parent PTMAEM.Cl, but also poor ability to undergo transcription following intranuclear injection, which may be due to the steric protection of the DNA from polymerases by the presence of HPMA. Finally, the influence of cationic polymer molecular weight on the properties of complexes formed with DNA was examined and there were some indications of the effects of molecular weight on transfection activity against HEK 293 cells for many of the polymers examined in this study. PVA.HCl, PMAEDA.HCl, PMAGEDA.HCl, and PTMAEM.Cl:DNA complexes formed with higher molecular weight cationic polymer often showed greater expression of reporter genes, which was usually also linked to an increased cytotoxicity.
2.1.2.2 With Steric Stabilizer
Most of the publications on polycations for DNA condensation possessing a steric stabilizer deal with the influence of the polymer architecture on the properties of the polyplexes (physico-chemical characteristics and transfection efficiency). Two types of architectures are mainly studied: linear copolymers with block and/or graft (eventually brush) architectures (Scheme 17). The steric stabilizers most commonly used are based on ethylene glycol or contain hydroxyl groups such as hydroxyethyl methacrylate or sugars (only a few examples are presented here because sugar-based polycations are out of the scope of this review).

Model cationic polymers containing different architectures of equivalent steric stabilizer components (represented by blue line) and comparable cationic components (represented by gray lines with positive charges). Reprinted with permission from [181]. Copyright 2011 Elsevier
2.1.2.2.1 PDMAEMA Derivatives
For PDMAEMA-b-PEG (Fig. 12a), PDMAEMA-b-POEGMA, and P(DMAEMA-stat-PEGMA) (Fig. 12b) of relatively low M n (7.8–21 kDa), the introduction of PEG did not significantly reduce the buffering capacity of PDMAEMA-based copolymers (for equivalent contents of DMAEMA units), but logically the buffering capacity is dependent on the DMAEMA content [182]. Of these polymers, the comb-type PDMAEMA-stat-PEGMA had the best complexing properties because charge neutrality was reached at a lower monomer:nucleotide molar ratio, which could be explained by its higher content of DMAEMA units compared to the other polymers (66% versus 30–37%). The steric effect of PEG chains prevented the addition of excess polymer to the polyplexes but did not seem to hinder the complexation/condensation with DNA and even seemed to have a beneficial effect. The performances of these PEG-containing copolymers were better than for the PDMAEMA homopolymer at monomer:nucleotide ratio above that needed for neutralization as regards complexation in the EtBr displacement assay as well as the condensation properties (as illustrated by the nanoparticle size). As pointed out by the authors, the presence of a highly hydrophilic and non-condensing PEG block in the copolymer may be expected to decrease the affinity for binding DNA due to an unfavorable entropy change [183]. However, enhanced binding may be connected to a local crowding effect of the PEG chain [184], or a decrease in the polarity of the polyion environment due to the presence of the PEG chains [185]. Moreover, the presence of PEG prevented the aggregation of polyplexes (as found in PDMAEMA polymer) and colloidally stable stoichiometric polyplexes were obtained. The capacity of a polymer to condense DNA is dependent on the DMAEMA content, but where the DMAEMA units are placed in the copolymers and the length of the PEG chains seem to play a role. Increasing DMAEMA content led to better condensing properties, i.e., smaller complexes. Despite, the propensity of these PEG-based copolymers to form small sterically stabilized complexes, their transfection ability in A549 cells at a monomer:nucleotide ratio inferior or equal to 5 was inferior to that of PDMAEMA homopolymer, maybe due to the presence of PEG, which provides a steric barrier around the polyplexes that inhibits contact with cells. In a subsequent publication [186], Stolnik and colleagues studied the cellular association and uptake and transfection efficiency of these copolymers at a fixed monomer:nucleotide molar ratio on three cell lines (A459, hepG2 and COS-7 cells) because transfection efficiency can be cell type-dependent. The transfection efficiencies were similar in the three cell lines for a given polymer but the rate of uptake of the complexes depended on the cell line used. Comb polymer P(DMAEMA-stat-PEGMA) consistently showed lower transfection efficiency than the linear PDMAEMA-b-PEG and bottle-brush PDMAEMA-b-POEGMA; the latter two giving similar transfection levels with all three cell lines. Indeed, P(DMAEMA-stat-PEGMA) showed less interaction with the cells (in flow cytometry studies) as well as less cell uptake (as shown by confocal microscopy) than the two other polymers. The authors explained this as being due to the different structures of the particles formed in the presence of DNA. Indeed, linear PDMAEMA-b-PEG and bottle-brush PDMAEMA-b-POEGMA have both diblock architectures, with the main difference being the spatial distribution of the EG units (linear PEG chain versus branched with OEG chains) but both polymers can form a micelle-like polyion complex with DNA. P(DMAEMA-stat-PEGMA) on the other hand, has several long pendant PEG chains (45 units) randomly distributed along the DMAEMA-based backbone; this statistical structure leads to rather soluble complexes (as shown by light scattering).

(a–d) Weak polycations with steric stabilizer: PDMAEMA derivatives
Given that a fraction of EG units much more than 20% in polymers seemed to hinder complexation in previous studies, Yang and colleagues conducted a systematic study [181] using a fixed fraction of EG units (~20%). This allowed comparison of various structures of PDMAEMA-PEG copolymers of the same molecular weight (M n ~ 11 kDa): block with linear PEG (PDMAEMA-b-PEG, Fig. 12a), brush block copolymer (PDMAEMA-b-POEGMA, Fig. 12b), and statistical copolymer P(DMAEMA-stat-OEGMA); for this last structure, the effect of molecular weight was also tested. All these polymers were found to complex well with DNA and completely retarded DNA migration at an N:P ratio of 2, showing that in this case (low fraction of EG), the structure of the PEG block did not affect the gene binding capacity of the polymers (nevertheless surprising for stat and brush copolymers). Similar results were obtained for polyanion exchange, but the difference can be seen in the capacity of DNA compaction, where the hydrodynamic diameter of the polyplexes was dependent both on molecular weight and structure of the copolymers but in general decreased with increasing N:P ratio for all polymers. Of the polymers with similar molecular weight but with different nature of PEGylation, the statistical copolymer was not effective at compacting DNA (micron-sized particles), whereas the two block-like polymers gave polyplexes of 100–200 nm in size (Fig. 13). The oligo-brush-like architecture led to smaller sized complexes than the simple diblock sequence, which might be due to the more compact structure of the shell (nevertheless not more resistant to heparin displacement) but their zeta potentials were similar. For the statistical copolymer series, the hydrodynamic sizes of the polyplexes dramatically decreased with increasing molecular weight at N:P ratio in the range 2–20, while the zeta potential stayed constant. The authors pointed out that molecular modeling studies on cationic polymer with neutral polymer grafts predicted that polymers of higher molecular weight were required to form smaller complexes [187]. Similar observations on the influence of short PEG grafts were also reported with PEI [188]. For cationic polymers with similar cationic charge but different nature of PEGylation, the diblock copolymer generally showed better transfection activity in HEK293 and HepG2 cells than the brush block copolymer or statistical copolymer with OEG chains.

TEM images of polymer/DNA complexes prepared in phosphate buffer (20 mM; pH = 6.5) at N:P 20 for (a) diblock copolymer PDMAEMA-b-PEG (x = 49, m = 47), (b) statistical copolymer P(DMAEMA-stat-OEGMA) (x = 46, y = 7), (c) statistical copolymer P(DMAEMA-stat-OEGMA) (x = 67, y = 9), (d) statistical copolymer P(DMAEMA-stat-OEGMA) (x = 90, y = 12) and (e) brush-block copolymer PDMAEMA-b-POEGMA (x = 53, y = 7, m = 8.5). Reprinted with permission from [181]. Copyright 2011 Elsevier
A study by Kataoka and colleagues of PDMAEMA-b-PEG (Fig. 12a), which had around the same ratio of DMAEMA:PEG as in the previous study but was twice as long [189], also showed the formation of micellar structures. There was a steep decrease in the size of these micelles with increasing N:P ratio, with a leveling off to approximately 95 nm in diameter at a N:P ratio of about 3, which was not only ascribed to DNA condensation (according to EtBr quenching, a fully condensed state occurred at N:P = 1.2) but also to a reduction in the association number to form non-stoichiometric micellar structures similarly to polyplexes based on PLL-b-PEG [190]. The presence of PEG in the polyplexes both increased the stability of pDNA against DNAse I and the affinity of the PDMAEMA segment for pDNA (exchange reaction with polyanions). Indeed, PEG provides a protecting layer by decreasing the permittivity of the microenvironment. The authors studied the transfection efficiency of this copolymer compared to PDMAEMA on HEK293 cells. In contrast to Stolnik and colleagues [182], they found a slightly better transfection efficiency of PDMAEMA-b-PEG compared to PDMAEMA, even at N:P ratio of 5 (and higher), possibly due to the longer chain length of the copolymer (or to the cell line). The authors also reported an unusual association of excess block copolymers on the micelles above the charge stoichiometric point, leading to an increase in zeta potential that finally leveled off at N:P = 15 (zeta potential of 17 mV), which could also be an explanation for the better transfection efficiency. Indeed, positively charged micellar polyplexes may benefit from a facilitated association with the cellular surface but, unfortunately, the comparison is difficult because the zeta potential was not reported in the studies of Stolnik and colleagues [182, 186]. Unfortunately, this improved performance was also associated with an increased toxicity compared to PDMAEMA.
For a similar PDMAEMA-b-PEG (Fig. 12a), Tam et al. took into consideration the critical micelle concentration (CMC; determined previously to be in the range 2 μg mL−1) of the polymer to explain the changes in the size and shape of the polyplexes (Fig. 14) [191]. The results corresponded to those of Kataoka, with a maximum condensation at N:P = 1.2 (EtBr quenching and zeta potential). As the polymer solution was added to the naked DNA (R h = 56 nm), at concentration below its CMC (shifted to higher values due to the presence of DNA), the polymer existed as a free cationic unimer that bound to the DNA to form complexes with a worm-like Gaussian structure. With the addition of more polymer, the polyplexes grew in size to around 90 nm (R h) probably due to secondary aggregation of the neutral nanoparticles (zeta potential). Above a N:P ratio of 1, the size of the polyplexes decreased and excess unbound polymeric unimers started to appear in the solution (DNA + 0.02 mg mL−1 polymer, i.e., N:P ratio of 2). At a polymer concentration above 0.06 mg mL−1, the polyplexes underwent significant structural rearrangements to form spherical aggregates of R h ≈ 35 nm, which was probably due to polymer aggregation above its CMC, accompanied by a coil–globule transition of the DNA molecules. The difference in the CMC with and without DNA can be attributed to two reasons. First, the DNA forms strong hydrogen bonds with water, which can “break” the water structure and consequently increase the CMC. Second, since binding between the polymer and DNA took place once the polymer solution was added, there were insufficient unbound polymer chains in the solution to induce micellization at 2 μg mL−1, shifting the CMC to a higher value [192].

(a) Proposed microstructure and R h of the DNA/PEG-b-PDEAEMA complex at various polymer concentrations in PBS solution. (b) TEM micrographs of DNA/PEG-b-PDEAEMA copolymer complex in PBS solution: 0.02 mg mL−1 polymer solution (upper part) and 0.2 mg mL−1 polymer solution (lower part). Reprinted with permission from [191]. Copyright 2006 American Chemical Society
A series of P(DMAEMA-co-OEGMA) (Fig. 12b) copolymers with constant chain length of 120 units but with various lengths of the OEG side chains and different comonomer compositions (variable DMAEMA/OEGMA composition, M n = 22.7–54.6 kDa) were studied by Rudolph and colleagues [193]. Among these polymers, DNA complexation by P(DMAEMA-co-OEGMA) copolymers with low f OEGMA (<50%) increased with increasing N:P ratios and decreased with increasing OEGMA molar ratio (f OEGMA) and molecular weight. At high f OEGMA (>50%), DNA complexation was abolished. Only at low f OEGMA (~17%) and low OEGMA molecular weight (about nine EG units) was pDNA migration completely retarded (indicating complete DNA complexation), as observed for PDMAEMA. Verbaan and colleagues had already observed that copolymers with a high degree of PEG grafting (i.e., PEG < 22%) were not capable of binding to pDNA [194]. In general, the efficiency of the polyplexes regarding complexation of pDNA decreases with increasing f OEGMA and OEGMA chain length, due to the reduction of the positive charge density of PDMAEMA and the introduction of sterically bulky OEGMA. This is consistent with the increasing particle size of the P(DMAEMA-co-OEGMA)-based polyplexes (condensation less efficient) and decreasing zeta potential (shielding effect of PEG) in a direct proportion to increasing f OEGMA and OEG chain length. None of these copolymers were cytotoxic to BEAS-2B cells, even at 500 μg mL−1. Following the same trend as for complexation, the transfection efficiency was dependent on the N:P ratio and decreased with increasing f OEGMA, in BEAS-2B cells as well as in MLE 12 cells. The transfection efficiency of the most efficient P(DMAEMA-co-OEGMA)-based polyplex was still more than 10 times lower than that of branched PEI25K/DNA, which can be explained by the inefficient pDNA condensation but also by reduced cellular uptake and endosomal escape.
PDMAEMA-b-POEGMA (Fig. 12b), PDMAEMA-b-POEGMA-b-PDMAEMA (Fig. 12c) with the same number of DMAEMA units (100), and POEGMA (66) form different self-assembled structures in solution both in the absence and presence of DNA due to their different architectures (diblock A-B or triblock A-B-A, Fig. 15c) [195]. The diblock completely retarded DNA migration at N:P ratio of ~0.6:1, whereas the triblock retarded migration at ~1.2:1 (at these ratios, the zeta potential was neutral and the polyplexes formed aggregates of ~1.8 μm, Fig. 15a, b). A similar trend was observed for the EtBr displacement, reflecting the better ability of the diblock to interact with pDNA. When the N:P ratio was more than 1.75:1, the particle sizes of the two polyplexes were almost the same (diameter of 200 nm) but the zeta potential of the triblock-based polyplexes was slightly lower (12 versus 17 mV), which could be explained by the larger content of POEGMA blocks at the surface (higher shielding effect) according to the structure that the authors proposed for the polyplexes (Fig. 15c). The triblock copolymer itself seemed to be far less toxic than the diblock to HEK293T cells (at 0.5 mg mL−1 there was nearly 100% cell viability versus 55% for the diblock). Following the trends of DNA complexation, the diblock copolymer was more efficient for transfection than the triblock at lower N:P ratio, with a bell-shaped dependence of the transfection efficiency as a function of the N:P ratio (Fig. 15d). For the range studied (N:P ratios of 1:1 to 15:), the maximum efficiency for PDMAEMA-b-POEGMA was reached at N:P = 3, while for PDMAEMA-b-POEGMA-b-PDMAEMA, the maximum seemed to be reached at N:P = 15, but it could also be a bell-shaped dependence shifted to higher ratios.

Dynamic light scattering results indicating the particle sizes (a) and zeta potentials (b) of the complexes as a function of N:P ratios for PDMAEMA-b-POEGMA-b-PDMAEMA/pDNA (triangles) and PDMAEMA-b-POEGMA/pDNA (dots). Data were obtained after the interaction of polymers with pDNA at various N:P ratios in TE buffer at pH 8.0. (c) Models for the formation of the PDMAEMA-b-POEGMA/pDNA and PDMAEMA-b-POEGMA-b-PDMAEMA/pDNA complexes. (d) Transfection efficiency determined by flow cytometry analysis of the GFP gene expression of PDMAEMA-b-POEGMA/pDNA and PDMAEMA-b-POEGMA-b-PDMAEMA/pDNA complexes in 293T cells as a function of N:P ratio [195]. Copyright 2011 Royal Society of Chemistry
PDMAEMA-b-PEG-b-PDMAEMA and PHEMA-b-PDMAEMA-b-PEG-b-PDMAEMA-b-PHEMA with 113 EG units in the central block and on each side 33, 48, or 61 DMAEMA units (used to complex DNA) and eventually ~15 more HEMA units on each side (Fig. 12d) were studied by Kang and colleagues [196]. At a polymer:plasmid weight ratio above 5, all polymers condensed DNA into particles of less than 200 nm in size and more than 25 mV in zeta potential. The triblocks PDMAEMA-b-PEG-b-PDMAEMA showed increased DNA complexing ability as well as transfection efficiency with increasing number of DMAEMA units. Adding short PHEMA blocks to the triblocks (leading to the pentablocks PHEMA-b-PDMAEMA-b-PEG-b-PDMAEMA-b-PHEMA) did not significantly impede the complexation ability of the polymers compared to the triblock and led to higher transfection than PDMAEMA. By comparison, a random block copolymer of similar component composition could not condense DNA efficiently, showing the importance of the architecture of the polycation.
2.1.2.2.2 Linear PEI Derivatives
PEG45-b-linear PEI600 (Fig. 16a) (M w = 51 kDa) was obtained by complete hydrolysis of a block copolymer PEG-b-poly(ethyl oxazoline-co-methyl oxazoline) [197]. PEG45-b-linear PEI600 inhibited DNA migration at a polymer:DNA weight ratio of 1 under physiological ionic strength, while linear PEI of 22 kDa achieved it at a ratio of 0.75. The levels of reporter gene expression obtained with the diblock copolymer were similar to those obtained for the linear PEI although at higher weight ratio, while the cytotoxicity as well as solubility were comparable, which was probably due to the relatively short length of the PEG block. Therefore, perhaps a longer PEG block should be introduced in order to see the benefits.

(a–c) Weak polycations with steric stabilizer: linear PEI derivatives
PHEMA-g-(linear PEI-b-PEG) (Fig. 16b) (M w = 509 kDa) adopted a cylindrical brush topology at pH 5.0 [198] and completely retarded DNA migration at N:P ratio of 2, whereas efficient condensation started at an N:P ratio of 10, leading to small particles of 150 nm in diameter with a positive zeta potential of 20 mV. PHEMA-g-(linear PEI-b-PEG) was well tolerated by HEK293 cells up to a dose of 125 μg mL−1, whereas exposure of the cells to PEI25K (branched) led to 50% viability at 16 μg mL−1. PHEMA-g-(linear PEI-b-PEG)/pDNA complexes were internalized by BT474 cells to a greater extent than PEI25K/DNA complexes at the same ratio (N:P = 10) and led to a higher transfection efficiency in a variety of cell lines.
POEGMA-b-(PHEMA-g-linear PEI) (Fig. 16c) is mainly composed of OEGMA units (89), which are thermoresponsive, and HEMA units grafted with linear PEI (3 or 8), which introduce more hydrophilicity and thus increase the LCST of the copolymer to close to body temperature [199]. Below their LCST, the polymer with most linear PEI grafts (8) retarded the movement of DNA at lower polymer:DNA ratio than the polymer with 3 grafts (3 versus 5). At 37°C, both polymers retarded DNA migration at a ratio of 3, given that the conformation of POEGMA89-b-(PHEMA-g-linear PEI)8 was unchanged, while at 37°C, i.e., 5°C higher than the LCST of POEGMA89-b-(PHEMA-g-linear PEI)3, the collapsed macromolecular chains covered more DNA and charges in the condensates, as confirmed by the zeta potential results. Moreover POEGMA89-b-(PHEMA-g-linear PEI)3 compacted DNA more tightly at 37°C than at 20°C (EtBr displacement assay). At a polymer:DNA ratio of 15, the polyplexes based on this last polymer reached a size acceptable for gene delivery (<200 nm) and the transfection efficiency was found to increase with increasing charge ratio both in COS-7 and HEK293 cells. At charge ratios of 25:1, POEGMA89-b-(PHEMA-g-linear PEI)3-based polyplexes achieved 100-fold higher transfection efficiency than PEI 1.2 kDa and levels comparable to that of PEI25K, while being far less toxic. As the incubation with DNA is usually done at 37°C, it is interesting to have non-viral gene delivery vectors that are more efficient at this temperature.
2.1.2.2.3 PLL Derivatives
At mixing charge ratio N:P of 1 [190], the polydispersity of the complexes of PEG272-b-PLL7, 19, 48 (Fig. 17a) with pDNA decreased dramatically before increasing again, whereas nearly complete condensation seemed to occur at N:P of 2:1 (EtBr exclusion assay), where the diameter of all the polyplexes was inferior to 100 nm. Note that condensation of a linearized pDNA by the same polymer was effective at lower N:P ratio. The explanation given by the authors is that native pDNA is in a super-coiled circular form, which certainly has a higher molecular restraint than the linear-formed DNA; thus, it is likely that the differences in molecular topology may crucially affect the condensation process of the DNA molecules in the sense that condensation may not be complete at a stoichiometric charge ratio for pDNA due to steric reasons, requiring excess PLL strands to promote further condensation. They also hypothesized that such a significant decrease in the average diameter between ratios 1 and 2 (~120 nm to ~90 nm) could be also due to a concomitant decrease in the association number of these micelle-like polyplexes, which was confirmed in the case of PEG272-b-PLL48 by LS. They could also show that a higher PEG content in the polymer resulted in micellar polyplexes with a decreased association number. The transfection efficiency in HEK293 cells was improved by increasing the length of the PLL segment and showed a bell-shaped dependency (as a function of the N:P ratio). The performance of PEG272-b-PLL48 was comparable to that of Lipofectamine™ and was partly attributed to a more favorable cellular association than that of the derivatives with a shorter PLL segment.

(a–e) Weak polycations with steric stabilizer: PLL derivatives
PLL-g-PEG (Fig. 17b), PLL-g-PHPMA (Fig. 17c), and PLL-g-dextran (Fig. 17d) were compared to PLL as transfection agents [185]. All these polymers were slightly hampered in their ability to condense DNA as compared to PLL (as shown by EtBr quenching), probably due to steric hindrance and charge screening by the hydrophilic blocks. PLL-g-PHPMA was the most effective of these new polymers despite its intermediate value for mass per charge (between that of PEG and dextran derivatives), which was not further explained. All these grafted copolymers with hydrophilic groups were capable of producing complexes with DNA that were more soluble than the PLL alone; as expected, the most efficient was the PEG-b-PLL with the longest PEG segment (12 kDa) at the highest grafting ratio (10%) and it was also the least toxic. PLL-b-dextran showed increased toxicity compared to low molecular weight PLL.
Polyoxazolines are thought to be an alternative to PEG as biocompatible blocks. In this frame, PLL-b-PMOx (Fig. 17e) and the polyplexes made thereof were synthesized and studied by the group of Lühmann [200]. As for PEG or other block copolymers comprising a cationic block and some hydrophilic modification, the complexation of DNA by PLL-b-PMOx was compromised at high grafting rates and/or long PMOx chains (as shown by gel electrophoresis). Condensates made of polymers with less than 7% grafting density showed aggregate formation at ratios supposedly above the neutral point (zeta potential not reported) and were over 500 nm in diameter. Above 7% grafting density, independent of the length of PMOx, the diameters of the polyplexes were less than 200 nm. These polymers were able to protect DNA from DNAse I digestion, but only PLL-g-PMOx with low molecular weight PMOx and low grafting densities of 7–14% showed significant gene expression in COS-7 cells (good transfection efficiency for this polymer was found at N:P ratio of 3.125), which correlated with their good cellular uptake.
2.1.2.2.4 Other Amino Acid-Based Polymers
PEG-co-(PLL-g-His) (Fig. 18a), a copolymer of PEG (1.45 kDa) and PLL modified with histidine at various grafting rates (5, 9, 16, and 22%) showed relatively poor EtBr displacement capacity [201]. EtBr displacement was only achieved at N:P ratio of 5 for the polymers with the two lowest grafting rates, and at ratios of 10 for the two others, which can be explained by a looser complexation due to the presence of PEG or the bulky imidazole groups or by the copolymer nature or by less cationic residues due to the grafting. Consistent with these high N:P ratios were the diameters of most of the complexes that remained between 150 and 200 nm for N:P ratios of 10 and above, whereas the complexes based on PLL were much smaller. The complexes of DNA based on PEG-co-(PLL-g-His16%) showed the highest transfection efficiency in the A7r5 cell line compared to the other polymers and the efficiency increased with the N:P ratio. Compared to polymers with lower degree of modification, this could be explained by the presence of more histidine residues, which are known to have endosomal buffering capacity (the buffering capacities increased with increasing His content), facilitating the escape of the polyplex into the cytoplasm. In addition, the decreased efficiency of PEG-co-(PLL-g-His22%) indicated the importance of having enough complexing units in the polymer. Probably because of the ester bonds, the polymer was totally degraded into its constituent PEG and PLL blocks after 24 h, which is an interesting approach to obtaining biodegradable polyplexes.

(a–e) Weak polycations with steric stabilizer: other amino acid-based polymers
PEG has also been coupled to polyamino acids other than PLL in order to increase solubility (critical in the case of polyhistidine for instance) and design a polymer with great DNA complexation and transfection properties.
PEG-b-PHis (Fig. 18b) and PHis-g-PEG (Fig. 18c) [202], would be presumably less able to complex and condense the genetic material into small polyplexes due to the relatively few protonated groups at pH 7 compared to the previous polymers, which had some free primary amine groups from the PLL segment [201]. In order to overcome this problem, the authors chose an approach where the complexation between the polymers and DNA was done at pH 5, at which the imidazole groups are protonated; then, the polyplexes were transferred into a neutral buffer, leading to partial deprotonation. The authors expected that DNA would remain in the partially protonated polyhistidine interior via hydrogen bonding and electrostatic interaction, while the partial deprotonation of polyhistidine would increase the hydrophobicity of the PHis segment, thus favoring the formation of micelles consisting of a DNA/PHis core surrounded by a PEG shell. The comb-shape polymer with 1 mol% ratio PEG complexed the DNA at a polymer:DNA weight ratio of 2, similarly to the block copolymer (PEG-b-PHis) and totally retained the DNA at a ratio of 3, while the other conjugates complexed DNA at a higher ratio and did not succeed in completely retaining DNA. This similarity between the comb-shape polymer and the block copolymer could eventually be explained by the low grafting rate, thus the effective structure of the polymers was rather similar. But, the polymer with the block structure, despite the low content of PEG compared to some of the comb-shape polymers, protected DNA from enzymatic degradation to the greatest extent, giving an indication that the structure in solution might be different. Nevertheless, it should be mentioned that even at high polymer:DNA ratio (4 and above), the charge of the polyplexes remained negative for all the polymers, probably reflecting the low protonation degree of histidine at pH 7. At pH 5, the polyplexes had diameters between 100 and 300 nm and, most importantly, were stable for over a week at room temperature. It could be observed that increasing the PEG content also increased the diameter of the polyplexes. Once transferred to pH 7, their stability was limited and aggregation occurred after 24 h, showing a certain limitation of this approach. Moreover, the transfection efficiency of all these polymers was poor in COS-7 cells (comparable to the tested PLL, but three orders of magnitude lower than PEI).
In contrast to PLL, polyaspartamide modified with chosen oligoethyleneimine side chains of various lengths had the advantage of possessing both primary and secondary amines. PEG-b-PAsp derivatives modified with various amines (Fig. 18d) were synthesized by the group of Kataoka [203]: the PAsp segment was modified with ethylene diamine [PEG-b-P(Asp-EDA), Fig. 18d-R 1], diethylene triamine [PEG-b-P(Asp-DETA), Fig. 18d-R 2], 4-methyldiethylene triamine [PEG-b-P(Asp-MDETA), Fig. 18d-R 3], and N,N-diethyldiethylene triamine [PEG-b-P(Asp-DEDETA), Fig. 18d-R 4]. The focus was on PEG-b-P(Asp-DETA), which showed a two-step protonation process (pK a 6.0 and 9.5) due to the presence of the ethylene diamine moiety, as illustrated in Fig. 19. At pH 7.4, this group is in the mono-protonated state (gauche form) and is capable of exerting a substantial buffering effect in the pH range down to 5. At pH 5, where 95% of the ethylene diamine unit is protonated (~diprotonated) the fluorescence in the EtBr dye-exclusion assay leveled off at N:P ratio of 1, while at pH 7.4 where the mono-protonated form is present, a N:P ratio of 2 is necessary to obtain substantial quenching.

Two-step protonation of the ethylene diamine unit with a distinctive gauche-anti conformational transition
It is interesting to note that the diameter of the polyplex micelles stayed constant at around 70–90 nm throughout the range of N:P ratios (1–20), even at neutral zeta potential, showing the efficiency of the hydrophilic shell to prevent aggregation. The transfection efficiency of this polymer was compared to the other PAsp derivatives [PEG-b-P(Asp-EDA), PEG-b-P(Asp-MDETA), PEG-b-P(Asp-DEDETA)], which all showed comparable sizes and zeta potential to PEG-b-P(Asp-DETA). The polyplex based on the polymer modified with the 2-aminoethyl group (pK a 9.4), PEG-b-P(Asp-EDA), was far less efficient than PEG-b-P(Asp-DETA) (factor 103 at N:P ratio of 20), presumably because of the impaired buffering capacity of the polymer but also because of its marginal internalization by cells [203]. Concerning PEG-b-P(Asp-MDETA), which possesses a tertiary amine instead the secondary amine of PEG-b-P(Asp-DETA) (primary amine unchanged), and PEG-b-P(Asp-DEDETA), which possesses a tertiary amine instead of the primary amine of PEG-b-P(Asp-DETA) (secondary amine unchanged), they both showed lower transfection efficiency compared to PEG-b-P(Asp-DETA), especially at high N:P ratios, showing the importance of the presence of both primary and secondary amines in the polymers used for polyplex formation. Nevertheless, at comparable N:P ratio (10), this construct was less efficient by a factor of 5 than ExGen500™, but was also less cytotoxic. In order to improve the results of PEG-b-P(Asp-DETA) [203], Kataoka and colleagues introduced some lysine moieties, obtaining PEG-b-P[LLys-co-(Asp-DETA)] with various percentage of lysine units, i.e., 24, 47, 71, and 100% (Fig. 18e) [204]. As expected, the extent of fluorescence quenching in the EtBr dye-exclusion assay (i.e., tight complexation) was proportional to the amount of lysine units present in the polymer. Interestingly, the fluorescence, which was leveled off at a N:P ratio of 2 for PEG-b-P(Asp-DETA), showed a leveling off at N:P ratio of 1 for these PEG-b-P[LLys-co-(Asp-DETA)] (from ~25 to 100% lysine units), indicating the beneficial effect of the lysine units for complexation. This corresponded also to the zeta potential results, which were nearly neutral for all polymers at this N:P ratio. In view of the trend in the zeta potential, the authors suggested that at high concentration of polymer, the lysine units may preferentially bind to the pDNA, replacing the Asp-DETA units and resulting in the continuous binding of the block catiomers until the lysine units saturate the available binding sites. By replacing half or more of the Asp-DETA units by lysine units, the internalization of the polyplexes was increased tenfold and the transfection efficiency was 100 times that of PEG-b-P(Asp-DETA).
2.2 Amphiphilic Polycations
The cationic charges, which are needed for efficient DNA condensation, and hydrophobic domains, which promote membrane interaction, have been combined in hydrophobically modified polymers such as PEI, PLL, PAMAM, and poly(N-ethyl-4-vinylpyridinium) salts [205–208]. Moreover, the hydrophobic part contributes to the hydrophobically driven interaction of DNA with polycation [209]. This increases the hydrophobic component and therefore there is need of increased steric stabilization in order to obtain colloidally stable polyplexes. Some examples are presented in the next sections.
2.2.1 Strong Polyelectrolytes with Alkyl Chains
A pioneering work of Kabanov et al. dealt with poly(N-ethyl-4-vinylpyridinium) bromide (PEVP) and its copolymer with N-4-vinylpyridinium modified with a longer alkyl chain, PEVP-C (Fig. 20a) [207]. For mole ratio [PEVP]/[DNA] between 0 and 0.5, where the polycation was in excess, soluble non-stoichiometric polyelectrolyte complexes were formed and the polycations were uniformly distributed along the DNA molecules. Further addition of PEVP led to the formation of an insoluble component composed of PEC with higher PEVP content (disproportionation). The addition even at 3% of a cetyl chain to the polymer (DP w = 400) narrowed the mole ratio range where soluble PEC were formed to [PEVP-C]/[DNA] = 0–0.25, which did not go in the direction wanted for efficient polyplexes and gene delivery. Moreover, the cell membrane penetration properties of PEVP-C were less efficient than those of PEVP, thus showing that either these properties are not a simple function of hydrophobicity or, as suggested, the hydrophobic component was buried in the core of the polyplex.

(a–g) Amphiphilic polycations: strong polyelectrolytes with alkyl chains
Quaternized or partially quaternized derivatives of poly(4-vinylpyridine) (DP = 1,600, M w = 168 kDa) were synthesized by the group of Izumrudov [210]. Among them, four different series were synthesized: quarternized poly(4-vinylpyridine) with N-alkyl ester substituents (Cn-PVP, Fig. 20b), polycarboxybetaine with alkyl spacer (Cn-PCB, Fig. 20c), poly[(4-vinylpyridine)-co-(N-alkyl-4-vinylpyridinium)] and poly[(N-methyl-4-vinylpyridine)-co-(N-alkyl-4-vinylpyridinium)] both with various alkylation degree β (respectively Cn-PVP-β, Fig. 20d and Me-Cn-PVP-β, Fig. 20e). Unfortunately, relatively few comparisons between these polymers were presented in this publication regarding the physico-chemical characteristics of their polyplexes. At a charge ratio of 5, Cn-PVP-based polyplexes with short N-alkyl substituents (n = 1–3) remained relatively inefficient regarding transfection, whereas more efficiency was noticeable for n = 4 and 5 (which could be due to the more pronounced destabilizing properties of cell membranes), then decreased again for n = 6, which might be due to a certain hindrance of its interaction with DNA itself by the presence of the long hydrophobic chain. The substitution of the ester moiety in the side chain of Cn-PVP (n = 4 and 5) by a carboxylic group gave the corresponding polycarboxybetaines (Cn-PCB). After complexation with DNA at a charge ratio of 5, they showed far less transfection activity compared to the parent polycations. The presence of the carboxylic group certainly weakened the interaction with DNA and possibly required a higher charge ratio for complexation. For the Cn-PVP-β series with various quaternization degrees, at a charge ratio of 5 there was a bell-shaped dependency of the transfection efficiency as a function of the alkylation degree, with the maximum at β = 65% for n = 5 (1000% increase in efficiency compared to pDNA alone) (β = 40% for n = 6), despite the similar sizes and zeta potentials of the polyplexes over all the β range. The explanation of the authors regarding this increased transfection efficiency of the partially alkylated PVP, Cn-PVP-β, was the presence of the pyridine groups, which could eventually be protonated in acidic media and thus could play a role in the proton sponge effect. Interestingly, the further methylation of these Cn-PVP-β derivatives led to negligible transfection efficiencies.
More recently, amphiphilic dimethylaminopyridinium-containing polymethacrylates with tail-end geometries with octyl, dodecyl, and hexadecyl spacers (n = 8, 12, 16) neutralized by bromide (Br) and octylsulfonate (S8) counterions were studied (PnDMAP-X, Fig. 20f) [211]. This study allowed the comparison of pyridinium-based derivatives according to the length of the spacer, counterion, and geometry. These polymers possess two kinds of amino moieties: a tertiary amine tail linked directly to the heterocycle (not protonated under physiological conditions) and an ammonio group that forms part of the pyridinium heterocycle, which is involved in the electrostatic binding with DNA. The amphiphiles PnDMAP-X formed a mixture of worm-like and spherical micelles in water for concentrations above 0.5 mg mL−1, with P8DMAP-X forming the loosest structures. The weight ratios of PnDMAP-X:DNA needed to retard DNA in gel electrophoresis were about 1.5 for n = 8, 3–5 for n = 12, and 2.5–5 for P16DMAP-X for X = Br and 5–7.5 for X = S8. Thus, the decrease in charge density for these derivatives with increasing spacer length could not only be accounted for by this trend in the minimum charge ratio needed. The counterion effect appeared for n = 16, where a higher weight ratio for S8 than for Br is needed to complex DNA, probably due to the reduced accessibility of the pyridinium group for DNA with this alkyl counterion (due to its size and/or hydrophobicity). Moreover, the transfection efficiency as a function of the spacer length followed the same trend in both series (X = Br and S8), with the highest transfection efficiency being obtained for n = 12, followed by n = 8 and finally n = 16. Also, a bell-shaped dependency of the transfection efficiency was observed as function of the length of the spacer. As in the previous example, it seems that a compromise between membrane destabilization (also reflected by increased cytotoxicity) and efficient DNA complexation has to be found for this type of derivative.
Poly(1-vinylimidazole)s alkylated with various chain lengths (R) and quaternization degree β (PVIm-R-β, Fig. 20g) were studied by the group of Asayama [212]. Due to the fact that imidazole groups are negligibly charged at physiological pH, quaternary nitrogen atoms as strong electrolytes were introduced in the structure. In order to still benefit from the pH buffering capacity of imidazole groups at endosomal pH, the quaternization was only partial. Taking PVIm-Bu as an example, complete DNA retardation occurred at a ratio of [butylated imidazole]/[phosphate] of around 1. Given that the pK a of the unmodified imidazole groups is around 6, the efficiency of DNA complexation at pH 6 (retardation of DNA migration occurred at a ratio of [butylated imidazole]/[phosphate] of 0.5) was greater than at pH 7.4, benefiting from the protonation of these imidazole groups. Moreover, the polyplexes based on PVIm-Bu caused negligible hemolysis at pH 7.4 but had a membrane disruptive activity at endosomal pH, and their stability (against polyanion exchange) was between that of PVIm-NH2 and PVIm-Oct. Concerning the cytotoxicity, PVIm-R with short alkyl chains (methyl, ethyl, butyl) were relatively non-cytotoxic, while PVIm-Oct caused significant cytotoxicity. Gene transfection of the luciferase gene to Hep2 cells was dependent on the length and density of the alkyl chains: for PVIm-Bu; the higher the density of butylated imidazole groups, the better was the transfection efficiency at low charge ratios (+/− < 12). For a middle density of alkylated imidazole groups (~20%), PVIm-Me and PVIm-Et mediated a higher gene expression than PVIm-Bu even at lower +/− charge ratio. This could be explained, as in the last case, by too much screening of the charges with the butyl chains.
2.2.2 Amphiphilic Polymers and Lipopolymers
Lipopolymers are polymers containing lipid moieties such as a fatty acid or a steroid such as cholesterol. At least some of the polymers presented in this section could eventually form micelles due to their amphiphilic structure, but either the concentration of their solution is under the CMC or the micelles are diluted and/or destabilized during their addition to the DNA solution.
Hydrophobicity can be introduced onto the side chains with hydrophobic moieties such as cholesterol and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) as will be presented in the next two examples. P(QuatDMAEMA-co-Chol) (Fig. 21a) is a copolymer containing quaternary ammonium units and cholesterol but no tertiary amines [213]. As previously seen, the range where the polyplexes are stable is diminished if the content of hydrophobic groups is too high. By slow addition of polycation to DNA, at N:P ratio close to 1, flocculation was observed for polyplexes based on the polymers with the highest cholesterol content (6.3 and 8.7 mol%) and only negatively charged polyplexes could be prepared. By fast addition of the polycation with highest cholesterol content to DNA, the M w of the aggregates showed the inverse tendency and decreased with increasing N:P ratios, even close to unity as already observed in the studies by Kabanov and Kabanov [214] and Oupicky et al. [215], while staying relatively constant above N:P = 1 for the polyplexes based on the two other polycations (1.4 and 6.3 mol% cholesterol). Moreover, by fast addition of polycations to DNA, ζ-potential increased with increasing content of cholesterol, i.e., with the hydrophobicity of the polycations. This effect was explained by the authors as such: DNA and polycations were intermixed in the initially formed complex (electrostatic interactions) giving rise to hydrophobic particles. The particles started to attract hydrophobic polycations by hydrophobic interaction, forming core–shell structures with the strongly hydrophobic cholesterol moieties of polycations attached to the particle surface and the remaining positively charged parts of polycations forming the shell, increasing the ζ-potential of the particle surface. The particle growth was then stopped by repulsive interactions of positively charged PECs and polycations, and the colloid stability of PECs increased with increasing content of side chains bearing cholesterol moieties, which is related to the level of surface charge and hydrophobicity of polycations.

(a–f) Amphiphilic polycations: amphiphilic polymers and lipopolymers not forming micelles after addition to DNA
In the case of PEG-b-PLL-g-DOPE (Fig. 21b), the lengths of the PEG and PLL blocks were not given and the PEG-b-PLL precursor was relatively polydisperse [216]. The authors supposed that PEG-b-PLL-g-DOPE assembled into micelles, whose structure was then destroyed by addition of DNA (concentration before mixing not given). Unfortunately, the formation of micelles has not been proven (no TEM pictures of PEG-b-PLL-g-DOPE before complexation with DNA and no determination of CMC), but is extremely plausible given the concentration of DOPE in the copolymer. By addition of DNA, a structure composed of a PLL/DNA core with DOPE on the core surface and a hydrophilic PEG shell was proposed as model (Scheme 18b). With increasing the degree of modification with DOPE, the mass ratio of polycation to DNA needed to completely retard DNA migration increased: for the PEG-b-PLL precursor, the mass ratio was 6, and for PEG-b-PLL-g-DOPE polycations with degree of DOPE modification of 16, 30, and 56% it was 14, 20, and 25, respectively. The polyplexes based on these polymers showed improved transfection efficiency in HepG2 and HeLa cells compared to naked DNA and PEG-b-PLL/DNA and comparable results to PEI. The polymer with 30% modification with DOPE showed the best results, probably due to a good compromise between a lower degree of DOPE, which did not allow the complex to penetrate the membrane, and a too-high degree of DOPE, which reduced the ability of the polymer to complex DNA.

Formation of (a) polyion complex micelles (PIC) for PEG-b-PLL and (b) lipid-modified polyion complex micelles (LPCM) for PEG-b-PLL-g-DOPE. Reprinted with permission from [216]. Copyright 2012 Elsevier
In the last examples of this section, interesting chemical structures will be presented but unfortunately relatively little information regarding their physico-chemical characteristics and/or transfection efficiency is available. Poly[(2-aminoethyl vinyl ether)-co-(alkyl vinyl ether)], with various alkyl chain lengths such as methyl, ethyl, propyl, and butyl [P(Am-co-Me), Fig. 21c-R 1; P(Am-co-Et), Fig. 21c-R 2; P(Am-co-Prop), Fig. 21c-R 3; and P(Am-co-But), Fig. 21c-R 4, respectively) is a good example [217]. The membrane lytic activity of these polymers was dependent on the length of the alkyl chains: the longer the better. Optimal transfections activities were obtained at an N:P ratio of 4, with P(Am-co-But) being the most efficient (ten times more efficient than PEI under these conditions), which correlated with the membrane lytic activity.
Hydrophobicity can also be introduced via a degradable hydrophobic block in a copolymer such as polylactide. For instance, folate-P(EI-co-EtOz)-b-PLLA (Fig. 21d) was synthesized via the partial hydrolysis of poly(2-ethyl-2-oxazoline) block at 66% and more, and the folic acid moiety also contributed to the hydrophobicity of the construct [218]. Folate-P(EI-co-EtOz)-b-PLLA began to form complexes with DNA at a polymer:DNA ratio of 10, while linear PEI completely retarded DNA at a ratio of 6. Polymers containing a large amount of PLLA reduced toxicity on HeLa cells compared to linear PEI, but also mediated gene transfer less efficiently.
A non-degradable hydrophobic block in a copolymer can be used to introduce hydrophobicity. A good example is Pluronics and its hydrophobic segment poly(propylene glycol) (PPG), such as in PEG-PPG-PEG-b-PDMAEMA (Fig. 21e) [219]. The pK a value of PEG-PPG-PEG-b-PDMAEMA was 7.1, lower than PEG-b-PDMAEMA and PDMAEMA, due to the effect of Pluronic™ lowering the dielectric constant of the amino groups. The polymer possessed a CMC of 5 g L−1, which is relatively high. The polymer condensed DNA into polyplexes of 200 nm in diameter at polymer:DNA ratios of 6 and more and a slightly positive zeta potential. Compared to PEG-b-PDMAEMA, the condensation was less efficient but the transfection efficiency was much higher and at lower polymer:DNA ratio [220].
LPEI-b-PPG-b-LPEI (Fig. 21f) with various LPEI and PPG block lengths were studied [221]. Note that these polymers, at least the ones with the highest hydrophilic:hydrophobic ratio, may self-assemble into flower-like micelles. LPEI50-b-PPG36-b-LPEI50 and LPEI19-b-PPG36-b-LPEI19 were able to retard DNA migration at a polymer:DNA weight ratio of 3:4 and 1:1, respectively, while LPEI14-b-PPG68-b-LPEI14 was not able to retard DNA even at a ratio of 15:1. These data, in correlation with AFM studies, suggest that LPEI50-b-PPG36-b-LPEI50 forms micellar structures where the positive charges are still available for interactions with DNA, whereas in the case of LPEI19-b-PPG36-b-LPEI19, the positive charges must be buried in the structure, hindering efficient electrostatic interactions with DNA.
2.2.3 Micelles of Amphiphilic Polymers and Lipopolymers
The polymers presented in this section can form micelles due to their amphiphilic structure. Moreover, there is the possibility to use these micelles as multicarriers, with hydrophobic drug loaded in the hydrophobic interior of the micelles and the genetic material complexed on the positively charged shell, if the micelles can structurally resist the addition of DNA.
P(MDS-co-CES) (Fig. 22a) is a biodegradable copolymer with a polyester main chain and containing potentially hydrolytically labile urethano groups to link the cholesterol moieties [222]. Moreover, this polymer contains both quaternary ammonium groups (DNA binding) and tertiary amine groups (endosomal buffering). This polymer formed micelles (CMC = 1.9 mg mL−1), which were positively charged (72 mV) and had a diameter of 96 nm in sodium acetate buffer and these pre-formed micelles were used for complexation of pDNA. This approach is different from the approach previously seen, where the polymer was added to DNA and, consequently, micelle formation was hindered due to the stronger electrostatic interactions between DNA and the positively charged block of the copolymer. The obtained polyplexes exhibited decreased mobility in gel electrophoresis and complete retardation at N:P ratio of 2. By studying the changes in the microenvironment of pyrene entrapped in the micelles, the authors verified the integrity of the core–shell nanoparticles during the DNA binding process and that the DNA binding further increased the hydrophobicity of the pyrene’s microenvironment. The transfection efficiency of this nanocarrier was tested on HEK293, HepG2, and 4T1 mouse breast cancer cell lines and depended strongly on the cell type and the N:P ratio. In HepG2 cells, the uptake of nanoparticle/DNA-based complexes was higher than for PEI/DNA, possibly due to their higher positive charge, which at the same time probably hindered the release of DNA intracellularly, leading to slightly lower overall gene expression. Importantly the amount of nanoparticles needed for optimal gene transfection was much lower than their IC50 values (around 150 μg mL−1 depending on the cell line).

(a–c) Amphiphilic polycations: amphiphilic polymers and lipopolymers forming micelles even in the presence of DNA
The hydrophobic part in amphiphiles can be the backbone, for instance poly(ε-caprolactone) (PCL). Indeed, PCL-g-PDMAEMA (Fig. 22b) formed nanoparticles (CAC = 0.81 mg mL−1) in water with diameters of several hundreds of nanometers (probably vesicles) and zeta potential of more than 40 mV (Scheme 19) [223]. These nanoparticles were pH- and thermoresponsive due to the presence of the PDMAEMA; the NPs were in a swollen state at an acidic pH range 6.0–6.9 at 37°C or higher but retracted at pH 7.4 and became even smaller when the temperature was above 37°C. Complete retardation was observed for N:P ratio of 2. In this case, the environment of the pyrene did not become more hydrophobic during DNA binding. The polyplexes showed a bell-shaped dependency of the transfection efficiency as a function of N:P ratio in 293T cells, and comparable values to Lipofectamine™2000 at a N:P ratio of 10 and 15. The internalization of these polyplexes was relatively slow; adherence to the cell membrane arose after 6 h, and after 24 h internalization and release into the cytoplasm had taken place.

Preparation of PCL-g-PDMAEMA NPs with payloads of hydrophobic drugs and plasmid DNA [223]. Copyright 2011 Royal Society of Chemistry
Hydrophobicity can be introduced via hydrophobic amino acids such as phenylalanine, as in PEG-b-PLL-b-PLPhe (Fig. 22c), which formed micelles (25–45 nm in diameter). The CMC of PEG-b-PLL-b-PLPhe decreased with increasing hydrophobic content (i.e., phenylalanine units) [224]. These CMC values (>100 mg mL−1) are quite high compared to those of other polymers. The copolymers did not exhibit apparent toxicity until a concentration of 500 μg mL−1. Due to the presence of PLL, the micelles self-assembled from PEG-b-PLL-b-PLPhe possessed pH-sensitive properties: from pH 4 to 10 their hydrodynamic diameter decreased from 60 nm to 15 nm (Scheme 20). The weight ratios at which the polymers can condense DNA were 2 and 15 for the PEG-b-PLL-b-PLP containing 16 and 37% phenylalanine, respectively, due to the higher density of amino groups of the first polymer. Moreover, at a ratio of 20, the size of the polyplexes were respectively 250 nm and 650 nm, while the zeta potentials were 25 and −10 mV, showing the incomplete condensation of DNA with the polymer containing the most phenylalanine units. Their transfection efficiency at a ratio of 20 and more was less efficient than PEI at a ratio of 10, despite their capacity to be internalized by cells. It was nevertheless not clear in this study if PEG-b-PLL-b-PLPhe were still self-assembled as micelles for DNA delivery.

(a) Size distribution of PEG-b-PLL-b-PLPhe micelles at different pH values. (b) Self-assembly of PEG-b-PLL-b-PLPhe copolymers at different pH values [224]. Copyright 2011 Royal Society of Chemistry
3 Polyampholyte/DNA Complexes
Ampholytes are amphoteric molecules that contain both acidic and basic groups and exist mostly as zwitterions in a certain pH range. The pH at which the average charge is zero is known as the molecule’s isoelectric point.
3.1 Polyzwitterions
A zwitterion is a neutral molecule with a positive charge introduced via a cationic functional group such as quaternary ammonium or phosphonium, which bears no hydrogen atom, and with a negative electrical charge introduced via a functional group such as carboxylate, which may not be adjacent to the cationic site. The overall neutrality of the molecule arises via a kind of intramolecular acid–base reaction between, for instance, an ammonium and a carboxylate group. Good examples of zwitterions are phosphorylcholine and betaines.
3.1.1 Polycarboxybetaines
The complexation of DNA with poly(pyridinio carboxylate)s with various lengths of additional alkyl chain at the α carbon and a carboxylate always in β position (Fig. 23a), also called polycarboxybetaines (PCB), was studied by Izumrudov et al. [225]. Given that these PCB possess quaternary ammoniums that are charged at any pH, these polymers indeed proved to be soluble in the pH range 2–11. For the pH range where DNA remains in the native state (4.0–10.0) these PCB were not able to bind DNA strongly enough to squeeze out EtBr (~15% reduction in fluorescence at N:P ratio of 2), which was explained by the authors by the amino group and the carboxylate group in β position forming a rather stable ionic pair. The influence of the length of additional alkyl chain at the α carbon was negligible.

(a–e) Polyampholytes based on betaines
More interesting are poly(pyridinio carboxylate)s with various lengths of alkyl chain (m) between charges (PCB m , Fig. 23b) [226]. Even with increasing the length of the spacer between the quaternary ammonium and the carboxylate group (m = 1–8), a stable ion pair was formed in neutral and weakly acidic media, reflected by the hindered protonation of the carboxylate group (as shown by potentiometric titration), and could lead to a potential inhibition of their electrostatic interactions with DNA. Despite the similar potentiometric behavior of the PCB m series, the length of the spacer in these polybetaines had an influence on their complexation with DNA. At N:P ratio of 5, PCB5 showed a good propensity to exclude EtBr at pH 9 (reduction of 75% of fluorescence), followed by PCB2 (reduction of 15% in fluorescence), while PCB4 and PCB8 showed similar behaviors (less than 5% reduction in fluorescence). The polyplexes followed the same trend in the dissociation of the complex in presence of salt (NaCl). The authors suggested that the propensity of PCB m (for m = 3 and 4) to form betaine rings spontaneously in aqueous solutions may be responsible for the first minimum, whereas the second minimum could be attributed to the ability of the relatively long spacer in PCB8 for cross-binding intramolecular electrostatic interactions of the charges within neighboring repeat units.
From these results, it is clear that zwitterions can be used as steric stabilizer but that in order to complex DNA, positively charged moieties and a certain distance between the zwitterionic groups are needed. Other polybetaines with PDMAEMA or primary amines are presented in the next sections and constitute a further improvement of these systems.
3.1.2 Polyphosphobetaines
The zwitterionic 2-methacryloyloxyethyl phosphorylcholine (MPC) block is a highly hydrated structure, where each monomer associates with 12 water molecules [227, 228]. In a block copolymer, this block is thought to introduce steric stabilization for the polyplex after complexation of the polycationic block with DNA.
A series of PDMAEMA-b-PMPC (Fig. 23c) with various length of PMPC block starting from PDMAEMA40 or various lengths of PDMAEMA blocks starting from PMPC30 were studied by the group of Stolnik [229]. PDMAEMA40-based copolymers with shorter PMPC blocks (10 or 20) had DNA binding affinities comparable to PDMAEMA homopolymer and higher DNA binding affinities than copolymers with longer PMPC blocks. When the percentage of PMPC was 65% and higher, the copolymers exhibited decreased affinity for DNA, suggesting that the presence of a long PMPC block was deleterious to DNA complexation; moreover, above a 1:1 ratio, the presence of this steric stabilizer reduced the association of the excess polymer with the polyplexes formed, as already observed for PDMAEMA-b-PEG [182], and prevented aggregation, which was not the case for copolymers with shorter PMPC blocks (10 or 20). PDMAEMA40-b-PMPC30 and PDMAEMA40-b-PMPC40 formed well-defined polyplexes with hydrodynamic diameters of approximately 150 nm, while PDMAEMA40-b-PMPC50 formed larger polyplexes with DNA with higher polydispersity. In the PMPC30-based copolymers series, increasing the size of the PDMAEMA block resulted in higher condensation ability as well as in smaller polyplexes in the sub-200 nm size range (except for PDMAEMA10-b-PMPC30), but also decreased the solubility of the polyplexes. As expected, the presence of PMPC block reduced the cellular association of these polyplexes, which correlated with their low transfection efficiencies (in the range of free DNA). A content ratio of the MPC unit to tertiary amine higher than 2 was required to produce spherical, well-condensed particles; MPC unit to amine ratios lower than 2 produced irregular structures ranging from toroids to rods [230].
Narain and coworkers studied copolymers of N-(2-aminoethyl) methacrylamide and 2-methacryloxyethyl phosphorylcholine as block or statistical copolymers [PAEMA-b-PMPC, P(AEMA-stat-MPC)] and (3-aminopropyl) methacrylamide and 2-methacryloxyethyl phosphorylcholine as block or statistical copolymers [PAPMA-b-PMPC, P(APMA-stat-MPC)] (see Fig. 23d) [231] in the same range of molecular weights as in the previous study [229] and around 50% modification in MPC. Unfortunately, little information is given about the physico-chemical characteristics of the polyplexes. Moreover, it was not clear which polymer:DNA ratios were used in order to obtain polyplexes with diameters ranging from 50 to 200 nm, but in general statistical polymers yielded polyplexes with larger diameters and irregular shapes compared to the corresponding diblock copolymers (which yielded spherical nanoparticles). Hence, not only the composition of MPC-based copolymers had an influence on the size and shape of the polyplexes but, as already seen for other systems, the architecture also played an important role. Copolymers with low molecular weights (6–7 kDa) showed lower gene expression as compared to polymers with higher molecular weights (10–12 kDa). A further increase in molecular weight led to a decrease in gene expression, probably due to their higher cytotoxicity. Moreover, the block copolymer architecture resulted in better transfection efficiency than the statistical copolymer, which was not due to an enhanced cellular uptake.
3.1.3 Polysulfobetaines
Polymers based on a sulfobetaine and the triblock thereof, PSB-b-POEMA-b-PSB (Fig. 23e) were found to be able to complex DNA [232]. The approach relied on OEMA as steric stabilizer, while the zwitterionic sulfobetaine itself was supposed to complex DNA, despite the presence at a short distance of the quaternary ammonium in the middle of the chain and the sulfonate as chain end (electrostatic repulsion with DNA). Nevertheless, its potential interest relied in the schizophrenic behavior of the polymer: the poly{N-[3-(methacryloylamino)propyl]-N,N-dimethyl-N-(3-sulfopropyl) ammonium hydroxide}block possessing an UCST (as heat energy is required to dissociate the crosslinking points stemming from ion pairings between ammonium cation and sulfo anion), whereas the poly[2-(2-methoxyethoxy)ethyl methacrylate] possessed an LCST. In the series obtained at 1 wt%, the polymers possessed an UCST in the range 14–18°C, followed by an LCST in the range 22–24°C (both transitions are dependent on the length of the blocks and concentration of the polymers in solution, in the range of DP considered for the PSB block and for the POEMA block with DP of 160 and less). The homopolymer PSB200 quenched 75% of EtBr fluorescence at polymer:DNA ratio of 5 and completely quenched it at N:P ratio of 20. The polymer condensed DNA into nanoparticles of less than 80 nm in diameter. PSB50-b-POEMA100-b-PSB50 and PSB80-b-POEMA40-b-PSB80 both quenched 40–45% of the fluorescence at ratio of 5, and reached a plateau of 55–60% at a ratio of 20. At a ratio of 10, they formed NPs with average size of 120 nm that were less compact and with irregular shape. This superior quenching of EtBr fluorescence cannot only be explained by the distance between opposite charges if we refer to the case of polycarboxybetaine. The capacity of the copolymers to bind DNA decreased with a decrease in the relative proportion of the PSB block (decrease in charge density) as well as an increase in the proportion of POEMA (steric hindrance).
3.2 Polyamphoters
An amphoteric species is a molecule or ion that can react as an acid as well as a base. One type of amphoteric species are amphiprotic molecules, which can either donate or accept a proton; examples are amino acids and proteins, which have amine and carboxylic groups. The competition between the acid–base equilibria of these groups leads to additional “complications” in their physical behavior but can also present some advantages due to the variation of their charge as a function of pH.
Poly(1,2-propylene H-phosphonate) modified with spermidine (PPA-g-SP, Fig. 24a with m = 1) had a LD50 of 85 μg mL−1 in HEK293 and COS-7 cells [233]. Efficient complexation with DNA was achieved for N:P ratios of 1.5 and higher (as shown by gel electrophoresis). According to zeta potential measurements, the polyplexes reached neutrality at N:P ratio of ~3, where aggregation took place (>1 μm); at N:P ratio of 5, the polyplexes had a size of 250 nm and zeta potential of 10 mV, which reached a plateau of 25 mV at N:P ratio of 25. The transfection efficiency increased with the N:P ratio, reaching maximal transfection efficiency at N:P ratio between 15 and 20, where the transfection was 30 times higher than PLL-mediated transfection, but still 40 times lower than TransFast™–DNA complexes. Interestingly, the transfection efficiency of the PPA-g-SP/DNA complexes was a function (but not linear) of DNA dose and transfection time, as previously seen in other examples. The conditions of preparation (ionic strength) were also important: a fourfold increase in the DNA dose resulted in a 50-fold increase in transfection efficiency, while extension of the incubation time from 30 min to 2 h resulted in a two orders of magnitude increase in the transfection efficiency, but further incubation time did not show further improvement. Preparation of the complexes in 1 M NaCl resulted in substantially larger particles (>1.3 μm), which showed a threefold increase in luciferase expression compared to complexes prepared in water (no explanation). With a similar polymer, poly(1,2-propylene H-phosphonate) modified with dipropyltriamine (PPA-g-DPA, Fig. 24a with m = 0) with either a similar grafting degree of 50% and different molecular weights or a similar backbone length but with different grafting degrees, the authors could study the influence of the molecular weight as well as grafting degree [234]. In the results presented by Ren et al., the ratio needed to complex DNA was not given but the studies started at N:P ratio of 10; which makes it quite difficult to compare these results with those previously reported. For a grafting density of 50%, an increase in molecular weight of the polycation led to an increase in DNA compaction ability of the polymer and slight increase in the polyplex size (60–80 nm). An increase in the net positive charge density for a given chain length also led to an increase in compaction ability (EtBr quenching) and, correspondingly, in a decrease in size of the polyplex (from 67 to 32 nm for 0.49 and 0.94 net positive charge density, respectively). At similar N:P ratio (10 for instance), it was remarkable that these polyplexes were much smaller than poly(1,2-propylene H-phosphonate) modified with spermidine (which could be due to other conditions of preparation, as these are not mentioned). These polyplexes were relatively stable under physiological conditions as well as with time, except those based on the shortest polymer (10 kDa) with a net positive charge density of 0.49. The cellular uptake of these polyplexes was cell line-dependent. In HeLa cells, the polyplexes with the second highest zeta potential but by far the smallest size were preferentially taken up; in HEK293 cells, the difference in uptake was not significant when the M w of the polymers was higher than 25 kDa; in HepG2 cells, there was no significant difference, showing that the zeta potential and size were not determining factors in these last two cell lines. Nevertheless, the transfection efficiency dramatically increased in all three cell lines with increasing molecular weight and grafting rate. As in the previous case, the influence of the partially negatively charged polymer backbone was not shown.

(a–c) Polyamphoters
Poly(isobutylene-alt-maleic acid-g-oligoethyleneamine) grafted with diethylene triamine P(Mal-g-DET), tetraethylenepentamine P(Mal-g-TEP), and pentaethylenehexamine P(Mal-g-PEH) (Fig. 24b) were studied by Yang and coworkers [235]. Complete retardation was only observed for the polymers with the longest oligoethylene amine chains at N:P ratio of 10. The polyplexes based on P(Mal-g-TEP) and P(Mal-g-PEH) had a neutral zeta potential at N:P ratio of about 12 and were positively charged for higher N:P ratio, but their size stayed in the micrometer range until an N:P ratio of 16 for P(Mal-g-TEP) and 22 for P(Mal-g-PEH). This size was not adequate for gene delivery and could be explained by the difficulty in condensing the negatively charged DNA with the partially negatively charged backbone. Above these critical ratios, the nanoparticles obtained were in the range of 400–800 nm, P(Mal-g-TEP) being less efficient than P(Mal-g-PEH) for condensing DNA, which could be explained by a greater number of protonated amine groups in P(Mal-g-PEH). Due to the larger particle size and lower zeta potential of P(Mal-g-TEP) compared to P(Mal-g-PEH), the polyplexes based on the latter polymer were most efficient in transfecting cells, comparable to the performance of PEI in some cases. The transfection efficiency was dependent upon N:P ratio and also on the cell line but, most importantly, polyplexes based on P(Mal-g-PEH) were localized to a large extent in the nucleus after 8 h.
At pH 7.4, prevailing negatively charged amphoteric PAAs were found to be relatively cytotoxic [236], while positively charged PAAs were far less cytotoxic [237]. Among them, PAgma (Fig. 24c) possesses three ionizable groups: a strong acid (pK a = 2.3), a medium-strength base (pK a = 7.4) and a strong base (pK a > 12.1) [238, 239]. At pH 7.4, PAgma has an excess average positive charge of 0.55 per unit (not influenced by the molecular weight) and had negligible cytotoxicity and hemolytic properties (synonymous of membrane damage) up to concentrations of 7 and 15 mg mL−1, respectively. But, as argued by the authors, the lack of membrane damaging properties did not necessarily imply lack of interactions with membranes, which is of importance for intracellular trafficking properties. At pH 7.4, complete retardation of DNA was achieved at N:P ratio of 15 for intermediate molecular weights (M n = 4.8 kDa). The size of the polyplexes decreased with increasing molecular weight until M n = 10 kDa, being less than 200 nm for this M n and around 270 nm for a polymer with M n = 20 kDa. At pH 5, compared to the values at pH 7.4, the size of the polyplexes decreased markedly and the zeta potential values became slightly more positive, which was due to an increase in average excess positive charge per polymer unit at this pH. The same rule as before seemed to apply: the smallest size and comparatively highest zeta potential helped the polyplexes based on polymers with molecular weight of 7–10 kDa to transfect cells more efficiently than polyplexes based on polymers with higher or lower molecular weights.
4 Conclusion
This review has shown that the design of polycations for gene delivery must take into account a balance between protection of DNA versus loss of efficiency for DNA condensation and efficient condensation versus hindering of DNA release, and that parameters leading to transfection efficiency in vivo still need to be optimized. Indeed, if the IPEC are not stable enough, premature dissociation will occur before delivery of the genetic material at the desired place, resulting in low transfection efficiency; on the other hand, a complex that is too stable will not release the DNA, also resulting in low gene expression. To determine these properties, gel electrophoresis to test the DNA/polymer complexation, EtBr or polyanion displacement to test the affinity of a polymer for DNA, and DLS to determine the extent of DNA condensation, are well-adapted techniques.
Polymers without steric stabilizer components were abandoned relatively early due to the inherent cytotoxicity of the permanent charges (even if these facilitate cellular entry of the polyplexes) and the propensity to be easily destabilized and precipitate. Strong complexation can also mean difficulty of release of the genetic material and, consequently, low transfection efficiency. The presence of steric stabilizers in the polyplexes results in an increased solubility under physiological conditions, but the problem of finding the right balance between steric stabilization and shielding of charges (that lowers the affinity of the polymer for DNA) is nevertheless present. On the other hand, this steric barrier and the shielding of the charges help the polymer to protect DNA from nuclease attacks, i.e., limit protein adsorption.
Unfortunately, until now, most of the polyplexes (if not all) presented in the studies reviewed here need to be prepared at high N:P ratio (higher than 10 and often even much higher) in order to be comparable to the commercial polymeric gene delivery agents (usually prepared at N:P ratios of less than 10).
Unfortunately, in all these cases, there are nevertheless problems in defining clear structure–activity relationships and explaining the apparent discrepancies between the behavior of the polyplexes in vitro and their poor performance in vivo. It is highly probable that the actual morphology of the polyplexes deviates from the expected structure (core–shell, etc.).
Moreover, it should not be forgotten that, since gene therapy using oligonucleotides such as antisense oligodeoxynucleotides (ODN); short RNA molecules such as small interfering RNA (siRNA), micro-RNA (miRNA), and short hairpin RNA (shRNA); or a DNAzyme that leads to a reduction in target/protein activity [240] takes more and more importance, the study of the complexes of these oligomeric materials with polycations is of increasing interest. Moreover, the application of DNA–polymer complexes is not limited to gene therapy and they also find use as DNA vaccines and biosensors.
Some challenges remain concerning the synthesis and structure of these polymers; for instance, finding new biocompatible polymers other than PEG. Moreover, in order to define clear structure–function relationships, it is necessary to use new polymerization techniques to obtain well-defined materials rather than randomized polymers [241]. Similarly, more architecturally controlled macromolecules such as dendronized polymers appear to be promising prospects in the field of polycations for gene delivery [242].
Abbreviations
- AFM:
-
Atomic force microscopy
- Arg:
-
Arginine
- bp:
-
Base pair
- CAC:
-
Critical aggregation concentration
- CMC:
-
Critical micelle concentration
- CMV:
-
Cytomegalovirus
- COS (cells):
-
CV-1 (simian) cell line carrying the SV40 genetic material
- ctDNA:
-
Calf thymus DNA
- Da:
-
Dalton, g.mol−1
- DLS:
-
Dynamic light scattering
- DLVO:
-
Derjaguin, Landau, Verwey, and Overbeek theory
- DNA:
-
Deoxyribonucleic acid
- DP:
-
Degree of polymerization
- ds:
-
Double stranded
- EGFP:
-
Enhanced green fluorescent protein
- EM:
-
Electron microscopy
- EtBr:
-
Ethidium bromide
- Glu:
-
Glutamic acid
- HEK (cells):
-
Human embryonic kidney cell line
- HepG2 (cells):
-
Human hepatocarcinoma cell line with epithelial morphology
- His:
-
Histidine
- HIV:
-
Human immunodeficiency virus
- IPEC:
-
Interpolylectrolyte complex
- LPEI:
-
Linear polyethyleneimine
- LS:
-
Light scattering
- Luc:
-
Luciferase
- Lys:
-
Lysine
- MPC:
-
2-Methacryloxyethyl phosphorylcholine
- MPS:
-
Mononuclear phagocyte system
- NCP:
-
Nucleosome core particle
- NMR:
-
Nuclear magnetic resonance
- PAMAM:
-
Poly(amido amine)
- PCL:
-
Poly(ε-caprolactone)
- PDI:
-
Polydispersity index
- PDMAEMA:
-
Poly[(2-dimethylamino) ethyl methacrylate]
- pDNA:
-
Plasmid DNA
- PEC:
-
Polyelectrolyte complex
- PEG:
-
Poly(ethylene glycol)
- PEI:
-
Polyethyleneimine
- PHEMA:
-
Poly(2-hydroxy ethyl methacrylate)
- PHPMA:
-
Poly(2-hydroxy propyl methacrylate)
- PLL:
-
Poly(l-lysine)
- PLLA:
-
Poly(l-lactide)
- PMMA:
-
Poly(methyl methacrylate)
- PNIPAM:
-
Poly(N-isopropyl acrylamide)
- PPI:
-
Poly(propylene imine)
- PTMAEMA:
-
Poly[(N-trimethylammonium) ethyl methacrylate]
- PVP:
-
Poly(4-vinylpyridine)
- RNA:
-
Ribonucleic acid
- SV:
-
Simian virus
- TEM:
-
Transmission electron microscopy
References
Kundu PP, Sharma V (2008) Synthetic polymeric vectors in gene therapy. Curr Opin Solid State Mater Sci 12:89–102
Wong SY, Pelet JM, Putnam D (2007) Polymer systems for gene delivery–past, present, and future. Prog Polym Sci 32:799–837
Dubruel P, Schacht E (2006) Vinyl polymers as non-viral gene delivery carriers: current status and prospects. Macromol Biosci 6:789–810
Kabanov AV, Felgner PL, Seymour LW (eds) (1998) Self-assembling complexes for gene delivery. Wiley, New York
Shcharbin DG, Klajnert B, Bryszewska M (2009) Dendrimers in gene transfection. Biochemistry 74:1070–1079
Dufès C, Uchegbu IF, Schätzlein AG (2005) Dendrimers in gene delivery. Adv Drug Deliv Rev 57:2177–2202
Ainalem M-L, Nylander T (2011) DNA condensation using cationic dendrimers–morphology and supramolecular structure of formed aggregates. Soft Matter 7:4577–4594
Guillot-Nieckowski M, Eisler S, Diederich F (2007) Dendritic vectors for gene transfection. New J Chem 31:1111–1127
Paleos CM, Tsiourvas D, Sideratou Z (2007) Molecular engineering of dendritic polymers and their application as drug and gene delivery systems. Mol Pharm 4:169–188
Fischer W, Calderón M, Haag R (2010) Hyperbranched polyamines for transfection. Top Curr Chem 296:95–129
Zhou Y, Huang W, Liu J, Zhu X, Yan D (2010) Self-assembly of hyperbranched polymers and its biomedical applications. Adv Mater 22:4567–4590
Nakayama Y (2012) Hyperbranched polymeric “star vectors” for effective DNA or siRNA delivery. Acc Chem Res 45:994–1004
Mao S, Sun W, Kissel T (2010) Chitosan-based formulations for delivery of DNA and siRNA. Adv Drug Deliv Rev 62:12–27
Azzam T, Domb AJ (2005) Cationic polysaccharides for gene delivery. In: Amiji MM (ed) Polymeric gene delivery–principles and applications. CRC, Boca Raton, pp 279–299
Yudovin-Farber I, Eliyahu H, Domb AJ (2007) Cationic polysaccharides for DNA delivery. In: Friedmann T, Rossi J (eds) Gene transfer: delivery and expression of DNA and RNA, a laboratory manual. CSHL, Cold Spring Harbor, pp 507–513
Froehlich T, Wagner E (2010) Peptide- and polymer-based delivery of therapeutic RNA. Soft Matter 6:226–234
Osada K, Kataoka K (2006) Drug and gene delivery based on supramolecular assembly of PEG-polypeptide hybrid block copolymers. Adv Polym Sci 202:113–153
Kircheis R, Wightman L, Wagner E (2001) Design and gene delivery activity of modified polyethylenimines. Adv Drug Deliv Rev 53:341–358
Boussif O, Lezoualc’h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr JP (1995) A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: Polyethylenimine. Proc Natl Acad Sci USA 92:7297–7301
Kim SW (2007) Polylysine copolymers for gene delivery. In: Friedmann T, Rossi J (eds) Gene transfer: delivery and expression of DNA and RNA, a laboratory manual. CSHL, Cold Spring Harbor, pp 461–471
Turrin C-O, Caminade A-M (2011) Dendrimers as transfection agents. In: Caminade A-M (ed) Dendrimers. Wiley, New York, pp 413–435
Kubasiak LA, Tomalia DA (2005) Cationic dendrimers as gene transfection vectors: dendri-poly(amidoamines) and dendri-poly(propylenimines). In: Amiji MM (ed) Polymeric gene delivery–principles and applications. CRC, Boca Raton, pp 133–157
Smedt SCD, Demeester J, Hennink WE (2000) Cationic polymer based gene delivery systems. Pharm Res 17:113–126
Bertin A, Hermes F, Schlaad H (2010) Biohybrid and peptide-based polymer vesicles. Adv Polym Sci 224:167–195
Iatrou H, Frielinghaus H, Hanski S, Ferderigos N, Ruokolainen J, Ikkala O, Richter D, Mays J, Hadjichristidis N (2007) Architecturally induced multiresponsive vesicles from well-defined polypeptides. Formation of gene vehicles. Biomacromolecules 8:2173–2181
Ho PS, Carter M (2011) DNA structure: alphabet soup for the cellular soul. In: Seligmann H (ed) DNA replication-current advances. InTech, New York
Calladine CR, Drew HR, Luisi BF, Travers AA (1992) Understanding DNA, 3rd edn. Elsevier Academic, Amsterdam
Ghosh A, Bansal M (2003) A glossary of DNA structures from A to Z. Acta Crystallogr Sect D Biol Crystallogr 59:620–626
Strick TR, Allemand J-F, Bensimon D, Bensimon A, Croquette V (1996) The elasticity of a single supercoiled DNA molecule. Science 271:1835–1837
Lu Y, Weers B, Stellwagen NC (2002) DNA persistence length revisited. Biopolymers 61:261–275
Chen H, Meisburger SP, Pabit SA, Sutton JL, Webb WW, Pollack L (2012) Ionic strength-dependent persistence lengths of single-stranded RNA and DNA. Proc Natl Acad Sci USA 109:799–804
Geggier S, Vologodskii A (2010) Sequence dependence of DNA bending rigidity. Proc Natl Acad Sci USA 107:15421–15426
Geggier S, Kotlyar A, Vologodskii A (2011) Temperature dependence of DNA persistence length. Nucleic Acids Res 39:1419–1426
Kypr J, Kejnovska I, Renciuk D, Vorlickova M (2009) Circular dichroism and conformational polymorphism of DNA. Nucleic Acids Res 37:1713–1725
Vorlíčkováa M, Kypra J, Sklenář V (2005) Nucleic acids: spectroscopic methods. In: Worsfold P, Townshend A, Poole C (eds) Encyclopedia of analytical science, 2nd edn. Elsevier, Amsterdam, pp 391–399
Mirkin SM (2001) DNA topology: fundamentals. In: Encyclopedia of life sciences. Wiley, New York, pp 1–11
Alfredsson V (2005) Cryo-TEM studies of DNA and DNA-lipid structures. Curr Opin Colloid Interface Sci 10:269–273
Lyubchenko YL (2004) DNA structure and dynamics–an atomic force microscopy study. Cell Biochem Biophys 41:75–98
Fransz P, Hd J (2011) From nucleosome to chromosome: a dynamic organization of genetic information. Plant J 66:4–17
Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ (1997) Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389:251–260
Travers A (1993) DNA-protein interactions. Chapman and Hall, London
Dobrynina AV, Rubinstein M (2005) Theory of polyelectrolytes in solutions and at surfaces. Prog Polym Sci 30:1049–1118
Radeva T (2001) Physical chemistry of polyelectrolytes. Surfactant science. CRC, Boca Raton
Manning GS (1969) Limiting laws and counterion condensation in polyelectrolyte solutions I. Colligative properties. J Chem Phys 51:924–933
Rühe J, Ballauff M, Biesalski M, Dziezok P, Gröhn F, Johannsmann D, Houbenov N, Hugenberg N, Konradi R, Minko S, Motornov M, Netz RR, Schmidt M, Seidel C, Stamm M, Stephan T, Usov D, Zhang H (2004) Polyelectrolyte brushes. Adv Polym Sci 165:79–150
Israelachvili J (1985) Intermolecular and surface forces. Academic, London
Thünemann AF, Müller M, Dautzenberg H, Joanny J-F, Löwen H (2004) Polyelectrolyte complexes. Adv Polym Sci 166:19–33
Gohy JF, Varshney SK, Antoun S, Jerome R (2000) Water-soluble complexes formed by sodium poly(4-styrenesulfonate) and a poly(2-vinylpyridinium)-block-poly(ethyleneoxide) copolymer. Macromolecules 33:9298–9305
Kabanov AV, Bronich TK, Kabanov VA, Yu K, Eisenberg A (1996) Soluble stoichiometric complexes from poly(N-ethyl-4-vinylpyridinium) cations and poly(ethylene oxide)-block-polymethacrylate anions. Macromolecules 29:6797–6802
Harada A, Kataoka K (1995) Formation of polyion complex micelles in an aqueous milieu from a pair of oppositely-charged block copolymers with poly(ethylene glycol) segments. Macromolecules 28:5294–5299
Kakizawa Y, Kataoka K (2002) Block copolymer micelles for delivery of gene and related compounds. Adv Drug Deliv Rev 54:203–222
Voets IK, Keizer AD, Cohen Stuart MA (2009) Complex coacervate core micelles. Adv Colloid Interface Sci 147–148:300–318
Ilarduya CT, Sun Y, Düzgünes N (2010) Gene delivery by lipoplexes and polyplexes. Eur J Pharm Sci 40:159–170
Kabanov AV, Kabanov VA (1998) Interpolyelectrolyte and block ionomer complexes for gene delivery: physicochemical aspects. Adv Drug Deliv Rev 30:49–60
Davis KA, Matyjaszewski K (2002) Statistical, gradient, block, and graft copolymers by controlled/living radical polymerizations. Adv Polym Sci 159:1–13
Matyjaszewski K (ed) (2003) Advances in controlled/living radical polymerization. ACS symposium series, vol 854. American Chemical Society, Washington
Matyjaszewski K (ed) (2006) Controlled/living radical polymerization: from synthesis to materials. ACS symposium series, vol 944. American Chemical Society, Washington
Xu Y, Plamper F, Ballauff M, Müller AHE (2010) Polyelectrolyte stars and cylindrical brushes. Adv Polym Sci 228:1–38
Bakeev KN, Izumrudov VA, Kuchanov SI, Zezin AB, Kabanov VA (1992) Kinetics and mechanism of interpolyelectrolyte exchange and addition reactions. Macromolecules 25:4249–4254
Tang MX, Szoka FC Jr (1998) Structure of polycation-DNA complexes and theory of compaction. In: Kabanov AV, Felgner PL, Seymour LW (eds) Self-assembling complexes for gene delivery. Wiley, New York
Dias RS, Pais AACC, Miguel MG, Lindman B (2003) Modeling of DNA compaction by polycations. J Chem Phys 119:8150–8157
Sarraguça JMG, Dias RS, Pais AACC (2006) Coil-globule coexistence and compaction of DNA chains. J Biol Phys 32:421–434
Dragan ES, Mihai M, Schwarz S (2006) Polyelectrolyte complex dispersions with a high colloidal stability controlled by the polyion structure and titrant addition rate. Colloid Surf A 290:213–221
Gärdlund L, Wågberg L, Norgren M (2007) New insights into the structure of polyelectrolyte complexes. J Colloid Interface Sci 312:237–246
Hartig SM, Greene RR, Dikov MM, Prokop A, Davidson JM (2007) Multifunctional nanoparticulate polyelectrolyte complexes. Pharm Res 24:2353–2369
Philipp B, Dautzenberg H, Linow KJ, Koetz J, Dawydoff W (1989) Polyelectrolyte complexes - recent developments and open problems. Prog Polym Sci 14:91–172
Giorgetti L, Siggers T, Tiana G, Caprara G, Notarbartolo S, Corona T, Pasparakis M, Milani P, Bulyk ML, Natoli G (2010) Noncooperative interactions between transcription factors and clustered DNA binding sites enable graded transcriptional responses to environmental inputs. Mol Cell 37:418–428
Danielsen S, Varum KM, Stokke BT (2004) Structural analysis of chitosan mediated DNA condensation by AFM: influence of chitosan molecular parameters. Biomacromolecules 5:928–936
Bloomfield VA (1996) DNA condensation. Curr Opin Struct Biol 6:334–341
Ainalem M-L, Carnerup AM, Janiak J, Alfredsson V, Nylander T, Schillen K (2009) Condensing DNA with poly(amido amine) dendrimers of different generations: means of controlling aggregate morphology. Soft Matter 5:2310–2320
Carnerup AM, Ainalem M-L, Alfredsson V, Nylander T (2011) Condensation of DNA using poly(amido amine) dendrimers: effect of salt concentration on aggregate morphology. Soft Matter 7:760–768
Kabanov VA, Zezin AB (1984) Soluble interpolymeric complexes as a new class of synthetic polyelectrolytes. Pure Appl Chem 56:343–354
Kiriy A, Yu J, Stamm M (2006) Interpolyelectrolyte complexes: a single-molecule insight. Langmuir 22:1800–1803
Dautzenberg H, Rother G (2003) Response of polyelectrolyte complexes to subsequent addition of sodium chloride: time-dependent static light scattering studies. Macromol Chem Phys 205:114–121
Discher DE, Eisenberg A (2002) Polymer vesicles. Science 297(5583):967–973. doi:10.1126/science.1074972
Lo C-L, Lin S-J, Tsai H-C, Chan W-H, Tsai C-H, Cheng C-HD, Hsiue G-H (2009) Mixed micelle systems formed from critical micelle concentration and temperature-sensitive diblock copolymers for doxorubicin delivery. Biomaterials 30(23–24):3961–3970. doi:10.1016/j.biomaterials.2009.04.002
Dautzenberg H (1997) Polyelectrolyte complex formation in highly aggregating systems. 1. Effect of salt: polyelectrolyte complex formation in the presence of NaCl. Macromolecules 30:7810–7815
Michaels AS, Mir L, Schneider NS (1965) A conductometric study of polycation-polyanion reactions in dilute aqueous solution. J Phys Chem 69:1447–1455
Sukhishvili SA, Kharlampieva E, Izumrudov V (2006) Where polyelectrolyte multilayers and polyelectrolyte complexes meet. Macromolecules 39:8873–8881
Maskos M, Stauber RH (2011) Characterization of nanoparticles in biological environments. In: Comprehensive biomaterials, vol 3: Methods of analysis. Elsevier, Amsterdam, pp 329–339
Lebovka NI (2012) Aggregation of charged colloidal particles. In: Müller M (ed) Advances in polymer science. Springer, Berlin, doi: 10.1007/12_2012_171
Patchornik A, Berger A, Katchalski E (1957) Poly-L-histidine. J Am Chem Soc 79:5227–5230
Palchaudhuri R, Hergenrother PJ (2007) DNA as a target for anticancer compounds: methods to determine the mode of binding and the mechanism of action. Curr Opin Biotechnol 18:497–503
Hannon MJ (2007) Supramolecular DNA recognition. Chem Soc Rev 36:280–295
Mulligan RC (1993) The basic science of gene therapy. Science 260:926–932
Verma IM, Somia N (1997) Gene therapy – promises, problems and prospects. Nature 389:239–242
Edelstein ML, Abedi MR, Wixon J, Edelstein RM (2004) Gene therapy clinical trials worldwide 1989–2004 – an overview. J Gene Med 6:597–602
Edelstein ML, Abedi MR, Wixon J (2007) Gene therapy clinical trials worldwide to 2007 – an update. J Gene Med 9:833–842
Palmer DH, Young LS, Mautner V (2006) Cancer gene-therapy: clinical trials. Trends Biotechnol 24:76–82
Yeung ML, Bennasser Y, Le SY, Jeang KT (2005) siRNA, miRNA and HIV: promises and challenges. Cell Res 15:935–946
Kay MA, Glorioso JC, Naldini L (2001) Viral vectors for gene therapy: the art of turning infectious agents into vehicles of therapeutics. Nat Med 7:33–40
Behr J-P (1993) Synthetic gene-transfer vectors. Acc Chem Res 26:274–278
Glover DJ, Lipps HJ, Jans DA (2005) Towards safe, non-viral therapeutic gene expression in humans. Nat Rev 6:299–310
Kawakami S, Higuchi Y, Hashida M (2008) Nonviral approaches for targeted delivery of plasmid DNA and oligonucleotide. J Pharm Sci 97:726–745
Scholz C, Wagner E (2012) Therapeutic plasmid DNA versus siRNA delivery: Common and different tasks for synthetic carriers. J Control Release 161:554–565
Zamore PD, Tuschl T, Sharp PA, Bartel DP (2000) RNAi. Cell Res 101:25–33
Bonetta L (2004) RNAi: silencing never sounded better. Nat Methods 1:79–86
Pecot CV, Calin GA, Coleman RL, Lopez-Berestein G, Sood AK (2011) RNA interference in the clinic: challenges and future directions. Nat Rev Cancer 11:59–67
Wilson RW, Bloomfield VA (1979) Counterion-induced condensation of deoxyribonucleic acid. A light-scattering study. Biochemistry 18:2192–2196
Agarwal S, Zhang Y, Maji S, Greiner A (2012) PDMAEMA based gene delivery materials. Mater Today 15:388–393
Brissault B, Kichler A, Guis C, Leborgne C, Danos O, Cheradame H (2003) Synthesis of linear polyethylenimine derivatives for DNA transfection. Bioconjug Chem 14:581–587
Rungsardthong U, Ehtezazi T, Bailey L, Armes SP, Garnett MC, Stolnik S (2003) Effect of polymer ionization on the interaction with DNA in nonviral gene delivery systems. Biomacromolecules 4:683–690
Nimesh S, Aggarwal A, Kumar P, Singh Y, Gupta KC, Chandra R (2007) Influence of acyl chain length on transfection mediated by acylated PEI nanoparticles. Int J Pharm 337:265–274
Gabrielson NP, Pack DW (2006) Acetylation of polyethylenimine enhances gene delivery via weakened polymer/DNA interactions. Biomacromolecules 7:2427–2435
Tang GP, Zeng JM, Gao SJ, Ma YX, Shi L, Li Y, Too H-P, Wang S (2003) Polyethylene glycol modified polyethylenimine for improved CNS gene transfer: effects of PEGylation extent. Biomaterials 24:2351–2362
Brumbach JH, Lin C, Yockman J, Kim WJ, Blevins KS, Engbersen JFJ, Feijen J, Kim SW (2010) Mixtures of poly(triethylenetetramine/cystamine bisacrylamide) and poly(triethylenetetramine/cystamine bisacrylamide)-g-poly(ethylene glycol) for improved gene delivery. Bioconjug Chem 21:1753–1761
Allen MHJ, Green MD, Getaneh HK, Miller KM, Long TE (2011) Tailoring charge density and hydrogen bonding of imidazolium copolymers for efficient gene delivery. Biomacromolecules 12:2243–2250
Prevette LE, Kodger TE, Reineke TM, Lynch ML (2007) Deciphering the role of hydrogen bonding in enhancing pDNA-polycation interactions. Langmuir 23:9773–9784
Georgiou TK, Vamvakaki M, Patrickios CS (2004) Nanoscopic cationic methacrylate star homopolymers: synthesis by group transfer polymerization, characterization and evaluation as transfection reagents. Biomacromolecules 5:2221–2229
Deshpande MC, Garnett MC, Vamvakaki M, Bailey L, Armes SP, Stolnik S (2002) Influence of polymer architecture on the structure of complexes formed by PEG-tertiary amine methacrylate copolymers and phosphorothioate oligonucleotide. J Control Release 81:185–199
Luten J, van Nostrum CF, Smedt SCD, Hennink WE (2008) Biodegradable polymers as non-viral carriers for plasmid DNA delivery. J Control Release 126:97–110
Laga R, Carlisle R, Tangney M, Ulbrich K, Seymour LW (2012) Polymer coatings for delivery of nucleic acid therapeutics. J Control Release 161:537–553
Chan P, Kurisawa M, Chung JE, Yang Y-Y (2007) Synthesis and characterization of chitosan-g-poly(ethylene glycol)-folate as a non-viral carrier for tumor-targeted gene delivery. Biomaterials 28:540–549
Blessing T, Kursa M, Holzhauser R, Kircheis R, Wagner E (2001) Different strategies for formation of PEGylated EGF-conjugated PEI/DNA complexes for targeted gene delivery. Bioconjug Chem 12:529–537
Kursa M, Walker GF, Roessler V, Ogris M, Roedl W, Kircheis R, Wagner E (2003) Novel shielded transferrin-polyethylene glycol-polyethylenimine/DNA complexes for systemic tumor-targeted gene transfer. Bioconjug Chem 14:222–231
Nishikawa M, Huang L (2001) Non viral vectors in the new millennium: delivery barriers in gene transfer. Hum Gene Ther 12:861–870
Grigsby CL, Leong KW (2010) Balancing protection and release of DNA: tools to address a bottleneck of non-viral gene delivery. J R Soc Interface 7:S67–S82
Soliman M, Allen S, Davies MC, Alexander C (2010) Responsive polyelectrolyte complexes for triggered release of nucleic acid therapeutics. Chem Commun 46:5421–5433
Spain SG, Yasayan G, Soliman M, Heath F, Saeed AO, Alexander C (2011) Nanoparticles for nucleic acid delivery. In: Ducheyne P (ed) Comprehensive biomaterials, vol 4. Elsevier, Amsterdam, pp 389–410
Sonawane ND, Szoka FC Jr, Verkman A (2003) Chloride accumulation and swelling in endosomes enhances DNA transfer by polyamine DNA polyplexes. J Biol Chem 278:44826–44831
Hunter AC, Moghimi SM (2010) Cationic carriers of genetic material and cell death: a mitochondrial tale. Biochim Biophys Acta 1797:1203–1209
Izumrudov VA, Zhiryakova MV (1999) Stability of DNA-containing interpolyelectrolyte complexes in water-salt solutions. Macromol Chem Phys 200:2533–2540
Xu FJ, Chai MY, Li WB, Ping Y, Tang GP, Yang WT, Ma J, Liu FS (2010) Well-defined poly(2-hydroxyl-3-(2-hydroxyethylamino)propyl methacrylate) vectors with low toxicity and high gene transfection efficiency. Biomacromolecules 11:1437–1442
Ma M, Li F, Yuan Z-F, Zhuo R-X (2010) Influence of hydroxyl groups on the biological properties of cationic polymethacrylates as gene vectors. Acta Biomater 6:2658–2665
Hemp ST, Allen J, Green MD, Long TE (2012) Phosphonium-containing polyelectrolytes for nonviral gene delivery. Biomacromolecules 13:231–238
Zelikin AN, Putnam D, Shastri P, Langer R, Izumrudov VA (2002) Aliphatic ionenes as gene delivery agents: elucidation of structure-function relationship through modification of charge density and polymer length. Bioconjug Chem 13:548–553
Izumrudov VA, Wahlund P-O, Gustavsson P-E, Larsson P-O, Galaev IY (2003) Factors controlling phase separation in water-salt solutions of DNA and polycations. Langmuir 19:4733–4739
Andersson T, Aseyev V, Tenhu H (2004) Complexation of DNA with poly(methacryl oxyethyl trimethylammonium chloride) and its poly(oxyethylene) grafted analogue. Biomacromolecules 5:1853–1861
Nisha CK, Manorama SV, Ganguli M, Maiti S, Kizhakkedathu JN (2004) Complexes of poly(ethylene glycol)-based cationic random copolymer and calf thymus DNA: a complete biophysical characterization. Langmuir 20:2386–2396
Howard KA, Dash PR, Read ML, Ward K, Tomkins LM, Nazarova O, Ulbrich K, Seymour LW (2000) Influence of hydrophilicity of cationic polymers on the biophysical properties of polyelectrolyte complexes formed by self-assembly with DNA. Biochim Biophys Acta 1475:245–255
Kimura T, Yamaoka T, Iwase R, Murakami A (2002) Effect of physicochemical properties of polyplexes composed of chemically modified PL derivatives on transfection efficiency in vitro. Macromol Biosci 2:437–446
Licciardi M, Campisi M, Cavallaro G, Carlisi B, Giammona G (2006) Novel cationic polyaspartamide with covalently linked carboxypropyl-trimethyl ammonium chloride as a candidate vector for gene delivery. Eur Polym J 42:823–834
Caputo A, Betti M, Altavilla G, Bonaccorsi A, Boarini C, Marchisio M, Buttò S, Sparnacci K, Laus M, Tondelli L, Ensoli B (2002) Micellar-type complexes of tailor-made synthetic block copolymers containing the HIV-1 tat DNA for vaccine application. Vaccine 20:2303–2317
Itaka K, Ishii T, Hasegawa Y, Kataoka K (2010) Biodegradable polyamino acid-based polycations as safe and effective gene carrier minimizing cumulative toxicity. Biomaterials 31:3707–3714
Zhang M, Liu M, Xue Y-N, Huang S-W, Zhuo R-X (2009) Polyaspartamide-based oligo-ethylenimine brushes with high buffer capacity and low cytotoxicity for highly efficient gene delivery. Bioconjug Chem 20:440–446
Cavallaro G, Scirè S, Licciardi M, Ogris M, Wagner E, Giammona G (2008) Polyhydroxyethylaspartamide-spermine copolymers: efficient vectors for gene delivery. J Control Release 131:54–63
Liu M, Chen J, Cheng Y-P, Xue Y-N, Zhuo R-X, Huang S-W (2010) Novel poly(amidoamine)s with pendant primary amines as highly efficient gene delivery vectors. Macromol Biosci 10:384–392
Kim T-I, Kim SW (2011) Bioreducible polymers for gene delivery. React Funct Polym 71:344–349
Lin C, Blaauboer C-J, Timoneda MM, Lok MC, van Steenbergen M, Hennink WE, Zhong Z, Feijen J, Engbersen JFJ (2008) Bioreducible poly(amido amine)s with oligoamine side chains: synthesis, characterization, and structural effects on gene delivery. J Control Release 126:166–174
Du F-S, Wang Y, Zhang R, Li Z-C (2010) Intelligent nucleic acid delivery systems based on stimuli-responsive polymers. Soft Matter 6:835–848
Verbaan FJ, Crommelin DJA, Hennink WE, Storm G (2005) Poly(2-(dimethylamino)ethyl methacrylate)-based polymers for the delivery of genes in vitro and in vivo. In: Amiji MM (ed) Polymeric gene delivery-principles and applications. CRC, Boca Raton
van de Wetering P, Cherng J-Y, Talsma H, Crommelin DJA, Hennink WE (1998) 2-(dimethylamino)ethyl methacrylate based (co)polymers as gene transfer agents. J Control Release 53:145–153
van de Wetering P, Moret EE, Schuurmans-Nieuwenbroek NME, van Steenbergen MJ, Hennink WE (1999) Structure-activity relationships of water-soluble cationic methacrylate/methacrylamide polymers for nonviral gene delivery. Bioconjug Chem 10:589–597
Asayama S, Maruyama A, Cho C-S, Akaike T (1997) Design of comb-type polyamine copolymers for a novel pH-sensitive DNA carrier. Bioconjug Chem 8:833–838
Jiang X, Lok MC, Hennink WE (2007) Degradable-brushed pHEMA-pDMAEMA synthesized via ATRP and click chemistry for gene delivery. Bioconjug Chem 18:2077–2084
Dubruel P, Strycker JD, Westbroek P, Bracke K, Temmerman E, Vandervoort J, Ludwig A, Schacht E (2002) Synthetic polyamines as vectors for gene delivery. Polym Int 51:948–957
Hinrichs WLJ, Schuurmans-Nieuwenbroek NME, van de Wetering P, Hennink WE (1999) Thermosensitive polymers as carriers for DNA delivery. J Control Release 60:249–259
Oupicky D, Reschel T, Konak C, Oupicka L (2003) Temperature-controlled behavior of self-assembly gene delivery vectors based on complexes of DNA with poly(L-lysine)-graft-poly(N-isopropylacrylamide). Macromolecules 36:6863–6872
Cheng N, Liu W, Cao Z, Ji W, Liang D, Guo G, Zhang J (2006) A study of thermoresponsive poly(N-isopropylacrylamide)/polyarginine bioconjugate non-viral transgene vectors. Biomaterials 27:4984–4992
Putnam D, Gentry CA, Pack DW, Langer R (2001) Polymer-based gene delivery with low cytotoxicity by a unique balance of side-chain termini. Proc Natl Acad Sci USA 98:1200–1205
Chen DJ, Majors BS, Zelikin A, Putnam D (2005) Structure-function relationships of gene delivery vectors in a limited polycation library. J Control Release 103:273–283
Burckhardt G, Zimmer C, Luck G (1976) Conformation and reactivity of DNA in the complex with protein: IV. Circular dichroism of poly-l-histidine model complexes with DNA polymers and specificity of the interaction. Nucleic Acids Res 3:561–580
Benns JM, Choi J-S, Mahato RI, Park J-S, Kim SW (2000) pH-sensitive cationic polymer gene delivery vehicle: N-Ac-poly(L-histidine)-graft-poly(L-lysine) comb shaped polymer. Bioconjug Chem 11:637–645
Roufai MB, Midoux P (2001) Histidylated polylysine as DNA vector: elevation of the imidazole protonation and reduced cellular uptake without change in the polyfection efficiency of serum stabilized negative polyplexes. Bioconjug Chem 12:92–99
Kim TH, Ihm JE, Choi YJ, Nah JW, Cho CS (2003) Efficient gene delivery by urocanic acid-modified chitosan. J Control Release 93:389–402
Park JS, Han TH, Lee KY, Han SS, Hwang JJ, Moon D, Kim SY, Cho YW (2006) N-acetyl histidine-conjugated glycol chitosan self-assembled nanoparticles for intracytoplasmic delivery of drugs: endocytosis, exocytosis and drug release. J Control Release 115:37–45
Mishra S, Heidel JD, Webster P, Davis ME (2006) Imidazole groups on a linear, cyclodextrin-containing polycation produce enhanced gene delivery via multiple processes. J Control Release 116:179–191
Swami A, Aggarwal A, Pathak A, Patnaik S, Kumar P, Singh Y, Gupta KC (2007) Imidazolyl-PEI modified nanoparticles for enhanced gene delivery. Int J Pharm 335:180–192
Pichon C, Goncalves C, Midoux P (2001) Histidine-rich peptides and polymers for nucleic acids delivery. Adv Drug Deliv Rev 53:75–94
Midoux P, Pichon C, Yaouanc J-J, Jaffrès P-A (2009) Chemical vectors for gene delivery: a current review on polymers, peptides and lipids containing histidine or imidazole as nucleic acids carriers. Br J Pharmacol 157:166–178
Dekie L, Toncheva V, Dubruel P, Schacht EH, Barrett L, Seymour LW (2000) Poly-L-glutamic acid derivatives as vectors for gene therapy. J Control Release 65:187–202
Dubruel P, Dekie L, Schacht E (2003) Poly-L-glutamic acid derivatives as multifunctional vectors for gene delivery. Part A. Synthesis and physicochemical evaluation. Biomacromolecules 4:1168–1176
Dubruel P, Dekie L, Christiaens B, Vanloo B, Rosseneu M, Vandekerckhove J, Mannisto M, Urtti A, Schacht E (2003) Poly-L-glutamic acid derivatives as multifunctional vectors for gene delivery. Part B. Biological evaluation. Biomacromolecules 4:1177–1183
Pack DW, Putnam D, Langer R (2000) Design of imidazole-containing endosomolytic biopolymers for gene delivery. Biotechnol Bioeng 67:217–223
Ramsay E, Hadgraft J, Birchall J, Gumbleton M (2000) Examination of the biophysical interaction between plasmid DNA and the polycations, polylysine and polyornithine, as a basis for their differential gene transfection in-vitro. Int J Pharm 210:97–107
Blauer G, Alfassi ZB (1967) A comparison between poly-α, L-ornithine and poly-α, L-lysine in solution: the effect of a CH2 group on the side-chain on the conformation of the poly-α-amino acids. Biochim Biophys Acta 133:206–218
Morgan DML, Larvin VL, Pearson JD (1989) Biochemical characterisation of polycation-induced cytotoxity to human vascular endothelial cells. J Cell Sci 94:553–559
Reich Z, Ittah Y, Weinberger S, Minsky A (1990) Chiral and structural discrimination in binding of polypeptides with condensed nucleic acid structures. J Biol Chem 265:5590–5594
Mann A, Khan MA, Shukla V, Ganguli M (2007) Atomic force microscopy reveals the assembly of potential DNA “nanocarriers” by poly-L-ornithine. Biophys Chem 129:126–136
Lim Y-B, Kim C-H, Kim K, Kim SW, Park J-S (2000) Development of a safe gene delivery system using biodegradable polymer, poly[α-(4-aminobutyl)-L-glycolic acid]. J Am Chem Soc 122:6524–6525
Putnam D, Langer R (1999) Poly(4-hydroxy-L-proline ester): low-temperature polycondensation and plasmid DNA complexation. Macromolecules 32:3658–3662
Simon RJ, Kania RS, Zuckermann RN, Huebner VD, Jewell DA, Banville S, Ng S, Wang L, Rosenberg S, Marlowe CK, Spellmeyer DC, Tan R, Frankel AD, Santi DV, Cohen FE, Bartlett PA (1992) Peptoids: a modular approach to drug discovery. Proc Natl Acad Sci USA 89:9367–9371
Fowler SA, Blackwell HE (2009) Structure-function relationships in peptoids: recent advances toward deciphering the structural requirements for biological function. Org Biomol Chem 7:1508–1524
Zuckermann RN, Kodadek T (2009) Peptoids as potential therapeutics. Curr Opin Mol Ther 11:299–307
Murphy JE, Uno T, Hamer JD, Cohen FE, Dwarki V, Zuckermann RN (1998) A combinatorial approach to the discovery of efficient cationic peptoid reagents for gene delivery. Proc Natl Acad Sci USA 95:1517–1522
Lobo BA, Vetro JA, Suich DM, Zuckermann RN, Middaugh CR (2003) Structure/function analysis of peptoid/lipitoid: DNA complexes. J Pharm Sci 92:1905–1918
Ji W, Panus D, Palumbo RN, Tang R, Wang C (2011) Poly(2-aminoethyl methacrylate) with well-defined chain length for DNA vaccine delivery to dendritic cells. Biomacromolecules 12:4373–4385
Luo S, Cheng R, Meng F, Park TG, Zhong Z (2011) Water soluble poly(histamine acrylamide) with superior buffer capacity mediates efficient and nontoxic in vitro gene transfection. J Polym Sci A Polym Chem 49:3366–3373
Sun H, Gao C (2010) Facile synthesis of multiamino vinyl poly(amino acid)s for promising bioapplications. Biomacromolecules 11:3609–3616
Wolfert MA, Dash PR, Nazarova O, Oupicky D, Seymour LW, Smart S, Strohalm J, Ulbrich K (1999) Polyelectrolyte vectors for gene delivery: influence of cationic polymer on biophysical properties of complexes formed with DNA. Bioconjug Chem 10:993–1004
Venkataraman S, Ong WL, Ong ZY, Loo SCJ, Ee PLR, Yang YY (2011) The role of PEG architecture and molecular weight in the gene transfection performance of PEGylated poly(dimethylaminoethyl methacrylate) based cationic polymers. Biomaterials 32:2369–2378
Rungsardthong U, Deshpande M, Bailey L, Vamvakaki M, Armes SP, Garnett MC, Stolnik S (2001) Copolymers of amine methacrylate with poly(ethylene glycol) as vectors for gene therapy. J Control Release 73:359–380
Stolnik S, Illum L, Davis SS (1995) Long circulating microparticulate drug carriers. Adv Drug Deliv Rev 16:195–214
Kleideiter G, Nordmeier E (1999) Poly(ethylene glycol)-induced DNA condensation in aqueous/methanol containing low-molecular-weight electrolyte solutions. Part II. Comparison between experiment and theory. Polymer 40:4025–4033
Toncheva V, Wolfert MA, Dash PR, Oupicky D, Ulbrich K, Seymour LW, Schacht EH (1998) Novel vectors for gene delivery formed by self-assembly of DNA with poly(L-lysine) grafted with hydrophilic polymers. Biochim Biophys Acta 1380:354–368
Deshpande MC, Davies MC, Garnett MC, Williams PM, Armitage D, Bailey L, Vamvakaki M, Armes SP, Stolnik S (2004) The effect of poly(ethylene glycol) molecular architecture on cellular interaction and uptake of DNA complexes. J Control Release 97:143–156
Ziebarth J, Wang Y (2010) Coarse-grained molecular dynamics simulations of DNA condensation by block copolymer and formation of core-corona structures. J Phys Chem B 114:6225–6232
Petersen H, Fechner PM, Martin AL, Kunath K, Stolnik S, Roberts CJ, Fischer D, Davies MC, Kissel T (2002) Polyethylenimine-graft-poly(ethylene glycol) copolymers: influence of copolymer block structure on DNA complexation and biological activities as gene delivery system. Bioconjug Chem 13:845–854
Wakebayashi D, Nishiyama N, Itaka K, Miyata K, Yamasaki Y, Harada A, Koyama H, Nagasaki Y, Kataoka K, Harada A (2004) Polyion complex micelles of pDNA with acetal-poly(ethylene glycol)-poly(2-(dimethylamino)ethyl methacrylate) block copolymer as the gene carrier system: physicochemical properties of micelles relevant to gene transfection efficacy. Biomacromolecules 5:2128–2136
Itaka K, Yamauchi K, Harada A, Nakamura K, Kawaguchi H, Kataoka K (2003) Polyion complex micelles from plasmid DNA and poly(ethylene glycol)-poly(l-lysine) block copolymer as serum-tolerable polyplex system: physicochemical properties of micelles relevant to gene transfection efficiency. Biomaterials 24:4495–4506
Tan JF, Too HP, Hatton TA, Tam KC (2006) Aggregation behavior and thermodynamics of binding between poly(ethylene oxide)-block-poly(2-(diethylamino)ethyl methacrylate) and plasmid DNA. Langmuir 22:3744–3750
Wang C, Tam KC (2002) New insights on the interaction mechanism within oppositely charged polymer/surfactant systems. Langmuir 18:6484–6490
Uzgun S, Akdemir O, Hasenpusch G, Maucksch C, Golas MM, Sander B, Stark H, Imker R, Lutz J-F, Rudolph C (2010) Characterization of tailor-made copolymers of oligo(ethylene glycol) methyl ether methacrylate and N, N-dimethylaminoethyl methacrylate as nonviral gene transfer agents: influence of macromolecular structure on gene vector particle properties and transfection efficiency. Biomacromolecules 11:39–50
Verbaan FJ, Oussoren C, Snel CJ, Crommelin DJA, Hennink WE, Storm G (2004) Steric stabilization of poly(2-(dimethylamino)ethyl methacrylate)-based polyplexes mediates prolonged circulation and tumor targeting in mice. J Gene Med 6:64–75
Yao Y, Feng D-F, Wu Y-P, Ye Q-J, Liu L, Li X-X, Hou S, Yang Y-L, Wang C, Li L, Feng X-Z (2011) Influence of block sequences in polymer vectors for gene transfection in vitro and toxicity assessment of zebrafish embryos in vivo. J Mater Chem 21:4538–4545
Xu F-J, Li H, Li J, Zhang Z, Kang E-T, Neoh K-G (2008) Pentablock copolymers of poly(ethylene glycol), poly((2-dimethyl amino) ethyl methacrylate) and poly(2-hydroxyethyl methacrylate) from consecutive atom transfer radical polymerizations for non-viral gene delivery. Biomaterials 29:3023–3033
Brissault B, Kichler A, Leborgne C, Danos O, Cheradame H, Gau J, Auvray L, Guis C (2006) Synthesis, characterization, and gene transfer application of poly(ethylene glycol-b-ethylenimine) with high molar mass polyamine block. Biomacromolecules 7:2863–2870
Liu X-Q, Du J-Z, Zhang C-P, Zhao F, Yang X-Z, Wang J (2010) Brush-shaped polycation with poly(ethylenimine)-b-poly(ethylene glycol) side chains as highly efficient gene delivery vector. Int J Pharm 392:118–126
Yang J, Zhang P, Tang L, Sun P, Liu W, Sun P, Zuo A, Liang D (2010) Temperature-tuned DNA condensation and gene transfection by PEI-g-(PMEO2MA-b-PHEMA) copolymer-based nonviral vectors. Biomaterials 31:144–155
Tv E, Zwicker S, Pidhatika B, Konradi R, Textor M, Hall H, Lühmann T (2011) Formation and characterization of DNA-polymer-condensates based on poly(2-methyl-2-oxazoline) grafted poly(L-lysine) for non-viral delivery of therapeutic DNA. Biomaterials 32:5291–5303
Bikram M, Ahn C-H, Chae SY, Lee M, Yockman JW, Kim SW (2004) Biodegradable poly(ethylene glycol)-co-poly(L-lysine)-g-histidine multiblock copolymers for nonviral gene delivery. Macromolecules 37:1903–1916
Putnam D, Zelikin AN, Izumrudov VA, Langer R (2003) Polyhistidine-PEG: DNA nanocomposites for gene delivery. Biomaterials 24:4425–4433
Kanayama N, Fukushima S, Nishiyama N, Itaka K, Jang W-D, Miyata K, Yamasaki Y, Chung U-I, Kataoka K (2006) A PEG-based biocompatible block catiomer with high buffering capacity for the construction of polyplex micelles showing efficient gene transfer toward primary cells. ChemMedChem 1:439–444
Miyata K, Fukushima S, Nishiyama N, Yamasaki Y, Kataoka K (2007) PEG-based block catiomers possessing DNA anchoring and endosomal escaping functions to form polyplex micelles with improved stability and high transfection efficacy. J Control Release 122:252–260
Vuillaume PY, Brunelle M, Calsteren M-RV, Laurent-Lewandowski S, Begin A, Lewandowski R, Talbot BG, ElAzhary Y (2005) Synthesis and characterization of new permanently charged poly(amidoammonium) salts and evaluation of their DNA complexes for gene transport. Biomacromolecules 6:1769–1781
Thomas M, Klibanov AM (2002) Enhancing polyethylenimine’s delivery of plasmid DNA into mammalian cells. Proc Natl Acad Sci USA 99:14640–14645
Kabanov AV, Astafyeva IV, Chikindas ML, Rosenblat CF, Kiselev VI, Severin ES, Kabanov VA (1991) DNA interpolyelectrolyte complexes as a tool for efficient cell transformation. Biopolymers 31:1437–1443
Doody AM, Korley JN, Dang KP, Zawaneh PN, Putnam D (2006) Characterizing the structure/function parameter space of hydrocarbon-conjugated branched polyethylenimine for DNA delivery in vitro. J Control Release 116:227–237
Liu Z, Zhang Z, Zhou C, Jiao Y (2010) Hydrophobic modifications of cationic polymers for gene delivery. Prog Polym Sci 35:1144–1162
Juan AS, Letourneur D, Izumrudov VA (2007) Quaternized poly(4-vinylpyridine)s as model gene delivery polycations: structure-function study by modification of side chain hydrophobicity and degree of alkylation. Bioconjug Chem 18:922–928
Vuillaume PY, Brunelle M, Bazuin CG, Talbot BG, Begin A, Calsteren M-RV, Laurent-Lewandowski S (2009) Tail-end amphiphilic dimethylaminopyridinium-containing polymethacrylates for gene delivery. New J Chem 33:1941–1950
Asayama S, Hakamatani T, Kawakami H (2010) Synthesis and characterization of alkylated poly(1-vinylimidazole) to control the stability of its DNA polyion complexes for gene delivery. Bioconjug Chem 21:646–652
Filippov SK, Konak C, Kopeckov P, Starovoytova L, Spirkova M, Stepanek P (2010) Effect of hydrophobic interactions on properties and stability of DNA-Polyelectrolyte complexes. Langmuir 26:4999–5006
Kabanov AV, Kabanov VA (1995) DNA complexes with polycations for the delivery of genetic material into cells. Bioconjug Chem 6:7–20
Oupicky D, Konak C, Ulbrich K, Wolfert MA, Seymour LW (2000) DNA delivery systems based on complexes of DNA with synthetic polycations and their copolymers. J Control Release 65:149–171
Sun X, Liu C, Liu D, Li P, Zhang N (2012) Novel biomimetic vectors with endosomal-escape agent enhancing gene transfection efficiency. Int J Pharm 425:62–72
Wakefield DH, Klein JJ, Wolff JA, Rozema DB (2005) Membrane activity and transfection ability of amphipathic polycations as a function of alkyl group size. Bioconjug Chem 16:1204–1208
Wang C-H, Hsiue G-H (2005) Polymer-DNA hybrid nanoparticles based on folate-polyethylenimine-block-poly(L-lactide). Bioconjug Chem 16:391–396
Bromberg L, Deshmukh S, Temchenko M, Iourtchenko L, Alakhov V, Alvarez-Lorenzo C, Barreiro-Iglesias R, Concheiro A, Hatton TA (2005) Polycationic block copolymers of poly(ethylene oxide) and poly(propylene oxide) for cell transfection. Bioconjug Chem 16:626–633
Alvarez-Lorenzo C, Barreiro-Iglesias R, Concheiro A (2005) Biophysical characterization of complexation of DNA with block copolymers of poly(2-dimethylaminoethyl) methacrylate, poly(ethylene oxide), and poly(propylene oxide). Langmuir 21:5142–5148
Lidgi-Guigui N, Guis C, Brissault B, Kichler A, Leborgne C, Scherman D, Labdi S, Curmi PA (2010) Investigation of DNA condensing properties of amphiphilic triblock cationic polymers by atomic force microscopy. Langmuir 26:17552–17557
Wang Y, Gao S, Ye W-H, Yoon HS, Yang Y-Y (2006) Co-delivery of drugs and DNA from cationic core-shell nanoparticles self-assembled from a biodegradable copolymer. Nat Mater 5:791–796
Guo S, Qiao Y, Wang W, He H, Deng L, Xing J, Xu J, Liang X-J, Dong A (2010) Poly(ε-caprolactone)-graft-poly(2-(N, N-dimethylamino) ethyl methacrylate) nanoparticles: pH dependent thermo-sensitive multifunctional carriers for gene and drug delivery. J Mater Chem 20:6935–6941
Li Y-Y, Hua S-H, Xiao W, Wang H-Y, Luo X-H, Li C, Cheng S-X, Zhang X-Z, Zhuo R-X (2011) Dual-vectors of anti-cancer drugs and genes based on pH-sensitive micelles self-assembled from hybrid polypeptide copolymers. J Mater Chem 21:3100–3106
Izumrudov VA, Zelikin AN, Zhiryakova MV, Jaeger W, Bohrisch J (2003) Interpolyelectrolyte reactions in solutions of polycarboxybetaines. J Phys Chem B 107:7982–7986
Izumrudov VA, Domashenko NI, Zhiryakova MV, Davydova OV (2005) Interpolyelectrolyte reactions in solutions of polycarboxybetaines, 2: Influence of alkyl spacer in the betaine moieties on complexing with polyanions. J Phys Chem B 109:17391–17399
Ishihara K, Nomura H, Mihara T, Kurita K, Iwasaki Y, Nakabayashi N (1998) Why do phospholipid polymers reduce protein adsorption? J Biomed Mater Res A 39:323–330
Konno T, Kurita K, Iwasaki Y, Nakabayashi N, Ishihara K (2001) Preparation of nanoparticles composed with bioinspired 2-methacryloyloxyethyl phosphorylcholine polymer. Biomaterials 22:1883–1889
Lam JKW, Ma Y, Armes SP, Lewis AL, Baldwin T, Stolnik S (2004) Phosphorylcholine-polycation diblock copolymers as synthetic vectors for gene delivery. J Control Release 100:293–312
Chim YTA, Lam JKW, Ma Y, Armes SP, Lewis AL, Roberts CJ, Stolnik S, Tendler SJB, Davies MC (2005) Structural study of DNA condensation induced by novel phosphorylcholine-based copolymers for gene delivery and relevance to DNA protection. Langmuir 21:3591–3598
Ahmed M, Bhuchar N, Ishihara K, Narain R (2011) Well-controlled cationic water-soluble phospholipid polymer-DNA nanocomplexes for gene delivery. Bioconjug Chem 22:1228–1238
Dai F, Wang P, Wang Y, Tang L, Yang J, Liu W, Li H, Wang G (2008) Double thermoresponsive polybetaine-based ABA triblock copolymers with capability to condense DNA. Polymer 49:5322–5328
Wang J, Zhang P-C, Lu H-F, Ma N, Wang S, Mao H-Q, Leong KW (2002) New polyphosphoramidate with a spermidine side chain as a gene carrier. J Control Release 83:157–168
Ren Y, Jiang X, Pan D, Mao H-Q (2010) Charge density and molecular weight of polyphosphoramidate gene carrier: are key parameters influencing its DNA compaction ability and transfection efficiency. Biomacromolecules 11:3432–3439
Khan M, Beniah G, Wiradharma N, Guo XD, Yang YY (2010) Brush-like amphoteric poly[isobutylene-alt-(maleic acid)-graft-oligoethyleneamine)]/DNA complexes for efficient gene transfection. Macromol Rapid Commun 31:1142–1147
Ferruti P, Manzoni S, Richardson SCW, Duncan R, Pattrick NG, Mendichi R, Casolaro M (2000) Amphoteric linear poly(amido-amine)s as endosomolytic polymers: correlation between physicochemical and biological properties. Macromolecules 33:7793–7800
Franchini J, Ranucci E, Ferruti P, Rossi M, Cavalli R (2006) Synthesis, physicochemical properties, and preliminary biological characterizations of a novel amphoteric agmatine-based poly(amidoamine) with RGD-like repeating units. Biomacromolecules 7:1215–1222
Ferruti P, Franchini J, Bencini M, Ranucci E, Zara GP, Serpe L, Primo L, Cavalli R (2007) Prevailingly cationic agmatine-based amphoteric polyamidoamine as a nontoxic, nonhemolytic, and “stealthlike” DNA complexing agent and transfection promoter. Biomacromolecules 8:1498–1504
Cavalli R, Bisazza A, Sessa R, Primo L, Fenili F, Manfredi A, Ranucci E, Ferruti P (2010) Amphoteric agmatine containing polyamidoamines as carriers for plasmid DNA in vitro and in vivo delivery. Biomacromolecules 11:2667–2674
Strachan T, Read AP (1999) Human molecular genetics, 2nd edn. Wiley-Liss, New York, Chap 22
Gössl I, Shu L, Schlüter D, Rabe JP (2002) Molecular structure of single DNA complexes with positively charged dendronized polymers. J Am Chem Soc 124:6860–6865
Laschewsky A (2012) Recent trends in the synthesis of polyelectrolytes. Curr Opin Colloid Interface Sci 17:56–63
Schlick T, Hayes J, Grigoryev S (2012) Toward convergence of experimental studies and theoretical modeling of the chromatin fiber. J Biol Chem 287:5183–5191
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Bertin, A. (2013). Polyelectrolyte Complexes of DNA and Polycations as Gene Delivery Vectors. In: Müller, M. (eds) Polyelectrolyte Complexes in the Dispersed and Solid State II. Advances in Polymer Science, vol 256. Springer, Berlin, Heidelberg. https://doi.org/10.1007/12_2013_218
Download citation
DOI: https://doi.org/10.1007/12_2013_218
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-40745-1
Online ISBN: 978-3-642-40746-8
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)