
Abstract
Coffee is cultivated in more than 70 countries of the intertropical belt where it has important economic, social and environmental impacts. As for many other crops, the development of molecular biology technics allowed to launch research projects for coffee analyzing gene expression. In the 90s decade, the first expression studies were performed by Northern-blot or PCR, and focused on genes coding enzymes of the main compounds (e.g., storage proteins, sugars, complex polysaccharides, caffeine and chlorogenic acids) found in green beans. Few years after, the development of 454 pyrosequencing technics generated expressed sequence tags (ESTs) obviously from beans but also from other organs (e.g., leaves and roots) of the two main cultivated coffee species, Coffea arabica and C. canephora. Together with the use of real-time quantitative PCR, these ESTs significantly raised the number of coffee gene expression studies leading to the identification of (1) key genes of biochemical pathways, (2) candidate genes involved in biotic and abiotic stresses as well as (3) molecular markers essential to assess the genetic diversity of the Coffea genus, for example. The development of more recent Illumina sequencing technology now allows large-scale transcriptome analysis in coffee plants and opens the way to analyze the effects on gene expression of complex biological processes like genotype and environment interactions, heterosis and gene regulation in polypoid context like in C. arabica. The aim of the present review is to make an extensive list of coffee genes studied and also to perform an inventory of large-scale sequencing (RNAseq) projects already done or on-going.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


1 Introduction: Once Upon a Time – The Story of Gene Expression in Coffee Plants
Despite the economic importance of coffee in international market, the knowledge about coffee molecular biology, and particularly regarding gene cloning and expression, can be considered as relatively recent. The first coffee genes described in the literature correspond to the complementary DNA (cDNA) sequences of α-galactosidase (Zhu and Goldstein 1994) and metallothionein I-like protein (Moisyadi and Stiles 1995), the first being a Short Communication in Gene and the second, a Plant Gene Register in Plant Physiology. Both articles only reported the cloning of these cDNAs without analyzing the expression of corresponding genes in coffee tissues.
This was the situation when I just arrived in Nestlé-Tours Research and Development Centre to initiate a project aiming to identify genes involved in coffee cup quality. Based on all the researches describing the importance of storage proteins (particularly in cereals) in the quality of final products, our interest was logically focused first to characterize these proteins in coffee fruits. Then in 1999, we reported the first article describing the expression of csp1 (coffee storage protein) gene coding the 11S proteins accumulated during bean development (Rogers et al. 1999a). At that time, gene expression studies were always performed by Northern blot experiments requiring both high quantities of total RNAs and the preliminary cloning of studied genes in order to synthetize their corresponding radio-labelled DNA probes. This situation persisted until the beginning of the 2000s, and in 2004, there were only 1,570 nucleotide sequences and 115 proteins from coffee deposited in GenBank/EMBL databases.
Few years after, with the development of high-throughput sequencing techniques, the first coffee EST (“expressed sequence tag”) sequencing projects were realized, and in 2016 there were 35,153; 25,574; and 25,574 unigenes available in public databases for Coffea arabica, Coffea canephora, and Coffea eugenioides, respectively. Then, the development of real-time quantitative PCR (RT-qPCR) technology significantly accelerated the number of coffee gene expression studies. The access to these ESTs also permitted to set up a 15 K microarray (“PUCECAFE”) DNA chip which was used to perform the first large-scale expression analyses aiming to understand transcription networks in flowers, mature beans, and leaves of C. canephora, C. eugenioides, and C. arabica (Privat et al. 2011). The same chip was also used to analyze the leaf expression of homeologous genes in response to changing temperature between C. arabica and its two ancestral parents, C. canephora and C. eugenioides (Bardil et al. 2011).
Soon after came the next-generation Illumina RNA sequencing (RNAseq) method enabling to perform expression analyses of thousands of genes by in silico approaches. The first article using such techniques was published by Combes et al. (2013) who studied the transcriptome in leaves of C. arabica submitted to warm and cold conditions suitable to C. canephora and C. eugenioides, respectively. Since this work, numerous other RNAseq studies were published, and many others are actually ongoing. Using all these data, it is now possible to generate reference transcriptomes which should help us to identify candidate genes (CGs) correlated with agronomic and quality traits in coffee.
2 Coffee Gene Expression
2.1 Reference Genes for qPCR Experiments
Since the development of EST sequencing projects (for reviews, see Lashermes et al. 2008; de Kochko et al. 2010, 2017; and Tran et al. 2016), RT-qPCR experiments, using either SYBR Green fluorochrome or specific TaqMan probes, are nowadays used in routine to study coffee gene expression. In order to quantify the expression levels, these experiments require the use of endogenous reference genes (as internal controls) which must be previously validated for particular tissues (Bustin 2002; Bustin et al. 2009). In that sense, several articles were published to identify the best reference genes to be used in different coffee tissues and growth conditions.
The first were published in 2009 showing that GAPDH (coding the glyceraldehyde 3-phosphate dehydrogenase) and UBQ10 (coding ubiquitin) were stable reference genes for normalization of qPCR experiments in different tissues of C. arabica, particularly in leaves and roots under drought stress (Barsalobres-Cavallari et al. 2009; Cruz et al. 2009). These two genes are also the most suitable for data normalization when analyzing multiple or single stresses in leaves of C. arabica and C. canephora (Goulao et al. 2012). In another study, Fernandes-Brum et al. (2017a) showed that the most stable reference genes were AP47 (coding the clathrin adaptor protein medium subunit), UBQ, (ubiquitin 60S), RPL39 (ribosomal protein L39), and EF1α (elongation factor 1-alpha) in all tissues of C. arabica, while GAPDH and UBQ, together with ADH2 (class III alcohol dehydrogenase) and ACT (β-actin), were the most stable for all tissues of C. canephora.
When analyzing the caffeine biosynthetic pathway, Sreedharan et al. (2018) showed that GAPDH and UBQ were the reference genes presenting the lowest variability in leaves and developing endosperm of C. canephora between control samples and treatments with salicylic acid (SA), methyl jasmonate (MeJA), light exposure, and PEG, which permitted the quantification of xanthosine methyltransferase (NMT) coding genes. In fact, UBQ was commonly used as a reference gene to normalize expression studies during bean development (Salmona et al. 2008; Joët et al. 2009, 2010, 2014; Cotta et al. 2014; Dussert et al. 2018) as well as in other coffee tissues, such as leaves and flower buds (Marraccini et al. 2011, 2012; Vieira et al. 2013; Mofatto et al. 2016). Even though several studies reported that RPL39 was not the most accurate reference (Cruz et al. 2009; de Carvalho et al. 2013), this gene was also used as a reference to compare expression profiles of several genes in developing beans and also in different organs such as leaves, stems, branches, roots, and flowers (Lepelley et al. 2007, 2012a, b; Pré et al. 2008; Privat et al. 2008; Simkin et al. 2006, 2008; Bottcher et al. 2011).
On the other hand, GAPDH and UBQ appeared to be the less stable reference genes for transcript normalization in C. arabica hypocotyls inoculated with Colletotrichum kahawae (causing the coffee berry disease (CBD)), for which the use of IDE (coding insulin degrading enzyme) and β-Tub9 (coding β-tubulin) (Figueiredo et al. 2013) as references is recommended. In another study, de Carvalho et al. (2013) showed that GAPDH together with MDH (coding malate dehydrogenase) and EF1α can be used as reference genes in leaves and roots of C. arabica subjected to N-starvation and heat stress, while UBQ10 was the most suitable reference for salt stress treatments. Using RefFinder, a web-based tool integrating geNorm, NormFinder, and BestKeeper programs (Xie et al. 2012), Martins et al. (2017) showed that MDH (malate dehydrogenase) presented the highest mRNA stability to study leaf gene expression in both C. arabica and C. canephora species subjected to single or multiple abiotic stresses such as elevated temperature and CO2 concentration ([CO2]). In another work, Freitas et al. (2017) showed that the 24S (ribosomal protein 24S) and PP2A (protein phosphatase 2A) genes were the most suitable references to study expression in embryogenic and non-embryogenic calli, embryogenic cell suspensions, and somatic embryos at different developmental stages in C. arabica.
2.2 Gene Expression in Coffee Species
At the time of writing this review (I apologize if I forgot mentioning some studies), the number of genes for which expression studies have been carried out individually was around 700. Most of these studies were performed by RT-qPCR using specific primer pairs designed against coffee ESTs generated by sequencing projects. In a chronological order, the first project was the Nestlé and Cornell initiative which generated around 63,000 ESTs from six cDNA libraries from fruits and leaves (at different developmental stages) of C. canephora clones of the Indonesian Coffee and Cocoa Research Institute (ICCRI) (Lin et al. 2005). Next was the IRD project which led more than 10,400 ESTs also from fruits and leaves of C. canephora (Poncet et al. 2006). Finally, the “Brazilian Coffee Genome” Project (BCGP), coordinated by the UNICAMP [University of Campinas] and the Embrapa [Empresa Brasileira de Pesquisa Agropecuária]), produced more than 200,000 ESTs (Vieira et al. 2006; Mondego et al. 2011) from C. arabica (≈187,000), from C. canephora (≈15,500), and also from C. racemosa (≈10,500). In order to identify the maximum of genes, this project used 43 cDNA libraries; most of them were built from transcripts extracted from fruits and leaves at different developmental stages but also from different plant organs (flowers, roots) and tissues (calli, cell suspensions, etc.) subjected to various biotic (e.g., roots infected with nematodes, stems infected with Xylella spp., leaves infected with miner Leucoptera coffeella and rust fungus Hemileia vastatrix) and abiotic (e.g., suspension cells treated with NaCl and chemicals such as acibenzolar-S-methyl and brassinosteroids) stresses.
As reported in Tables 1, 2, and 3, most of these expression studies were performed in C. arabica (n ≈ 550 genes) and C. canephora (n ≈ 100 genes), with a repartition reflecting quite well the importance of C. arabica (59%) and C. canephora (41%) species in the worldwide coffee production (ICO 2020). These expression studies were more limited in other coffee species such as in C. racemosa (n = 25), C. eugenioides (n = 18), and C. liberica (n = 5). For 70 genes, expression analyses were performed on both C. arabica and C. canephora species. However, a limited number of studies (described in Sect. 2.3) analyzed gene expression simultaneously in C. arabica, C. canephora, and C. eugenioides using specific primers and qPCR for each homeolog in each species. Several articles also reported in silico gene expression profiles which were not confirmed by RT-qPCR (Table 4).
2.3 Coffee Gene Expression in C. arabica: A Tricky Case
Before discussing gene expression in coffee, it is important to remember that C. arabica (2n = 4× = 44) is an allotetraploid coffee species derived from a natural hybridization event between the two diploid (2n = 2× = 22) species C. canephora and C. eugenioides (Lashermes et al. 1999) which occurred approximately 10,000–50,000 years ago (Cenci et al. 2012). Consequently, the transcriptome of C. arabica is a mixture of transcripts expressed from homeologous genes harbored by its two sub-genomes, respectively, namely, CaCc (also referred as Ca) for C. canephora sub-genome and CaCe (also referred as Ea) for C. eugenioides sub-genome.
In the first attempt to analyze gene expression contributions of each sub-genome in C. arabica, Vidal et al. (2010) used qPCR coupled with allele-specific combination TaqMAMA-based method (Li et al. 2004) and developed a pipeline to find SNP (single nucleotide polymorphism) haplotypes of CaCc and CaCe homeologs in the ESTs of the BCGP. Of the 2069 contigs studied, these authors observed a biased expression for 22% of them, with 10% overexpressing CaCc homeologs and 12% overexpressing CaCe homeologs, therefore showing that the two sub-genomes do not contribute equally to the transcriptome of C. arabica. By analyzing gene ontology (GO), these authors also proposed that the CaCe sub-genome expressed genes of proteins involved in basal biological processes (such as those related to photosynthesis, carbohydrate metabolic processes, aerobic respiration, and phosphorylation). On the other hand, the CaCc sub-genome contributed to adjust Arabica expression (e.g., to biotic and abiotic stresses) through the expression of genes of regulatory proteins such as those related to hormone stimuli (mainly auxin), GTP signal transduction, translation, and ribosome biogenesis proteasome activity.
The 15 K “PUCECAFE” microarray (Privat et al. 2011) was also used to perform genome-wide expression study in order to analyze the effects of warm and cold temperatures on leaf gene expression of C. arabica and those of its two ancestral parents (C. canephora and C. eugenioides) (Bardil et al. 2011). Even though this global gene expression analysis did not allow determining the relative contributions of homeologs to the C. arabica leaf transcriptome, it revealed the existence of transcription profile divergences between the allopolyploid and its parental species that were greatly affected by growth temperature. Two other “in silico” analyses that studied the effects of warm vs. cold temperature in C. arabica were performed. The first one used SNP ratio quantification to monitor the relative expression of 13 homeologous gene pairs in five organs (cotyledons, young leaves, leaves, stems, and roots) in addition of warm/cold temperatures (Combes et al. 2012). No case of gene silencing or organ-specific silencing was detected, but 10 out of 13 sampled genes showed biased expression: 4 genes toward CaCe, 4 genes toward CaCc, and 2 genes toward CaCe or CaCc depending on the organ considered. In the second study, the effects of warm/cold temperatures on C. arabica leaf transcriptome were analyzed by RNA sequencing (Combes et al. 2013). The relative homeologous gene expression, assessed in 9,959 and 10,628 pairs of homeologs in warm and cold growing conditions, respectively, revealed that 65% of these genes had an equivalent expression level, while the rest (35%) showed biased homeologous expression. Although the warm and cold conditions were suitable for C. canephora or C. eugenioides parental species, respectively, neither sub-genome appeared preferentially expressed to compose the final transcriptome of C. arabica.
Because CaCc and CaCe sub-genomes of C. arabica have low sequence divergence (with an average difference for genes of only 1.3%) (Cenci et al. 2012), we can conclude that all the studies analyzing gene expression in C. arabica by “wet lab” approaches (e.g., Northern blot experiments for the most ancient and even RT-qPCR using primer pairs probably designed in highly conserved cDNA regions) quantify the transcripts expressed by both CaCc and CaCe sub-genomes.
However, few studies succeed in discriminating specifically the expression of CaCc and CaCe homeologs in C. arabica. All of them (described below) used the presence of SNPs or the small insertions and deletions (INDELs), for example, present in the 3′ and 5′ untranslated regions (UTRs), to design CaCc and CaCe primer pairs which permitted to identify homeologous differential expression (HDE) by qPCR. The first one concerned the expression of the CaWRKY1a (CaCc) and CaWRKY1b (CaCe) genes in C. arabica (Petitot et al. 2008, 2013) coding transcription factors known to be associated with plant defense responses to biotic and abiotic stresses (reviewed in Ülker and Somssich 2004; Eulgem 2006). In this species, both homeologs were concomitantly expressed in leaves and roots under all treatments (salicylic acid and infection by leaf rust [H. vastatrix] and root-knot nematode (RKN) Meloidogyne exigua), suggesting that they undergo the same transcriptional control.
A different situation was observed in C. arabica for the RBCS1 gene with the predominant expression of the homeolog CaCe (over the CaCc homeolog) in the leaves of non-introgressed (“pure”) cultivars such as Typica, Bourbon, and Catuaí (Marraccini et al. 2011), suggesting that specific suppression of RBCS1 CaCc expression occurred during the evolutionary processes that generated the C. arabica species. This situation fits with the concept of genome dominance (or genome expression dominance) for which the total expression of homeologs of a given gene in an allopolyploid is statistically the same as only one of the parents (Grover et al. 2012). However, RBCS1 CaCe and CaCc homeologs were co-expressed (with the same order of magnitude) in the leaves of C. arabica Timor hybrid HT832/2 used to create the IAPAR59; Tupi and Obabã cultivars of C. arabica, for example; as well as in Icatú which comes from a cross between C. canephora and C. arabica Bourbon. For all these “introgressed” Arabica cultivars, CaCc expression was always higher than CaCe. The existence of a bias in favor of CaCc homeologs suggests that one (or several) genetic factor of C. canephora species was introgressed in C. arabica together with the HdT (hybrid of Timor, a spontaneous hybrid between C. arabica and C. canephora) genes conferring resistance to leaf rust and activated (or unrepressed) the CaCc sub-genome.
In a work analyzing the effects of abiotic stress on the expression of genes of the mannitol biosynthesis pathway, de Carvalho et al. (2014) reported that the CaCc homeologs of CaM6PR (coding mannose-6-phosphate reductase), CaPMI (coding phosphomannose isomerase), and CaMTD (coding the NAD+-dependent mannitol dehydrogenase, oxidizing mannitol to produce mannose) were also highly expressed in leaves of C. arabica IAPAR59 subjected to drought, high salinity, and heat-shock stress.
HDE was also observed when analyzing expression of nsLTP (encoding non-specific lipid transfer proteins) genes in the separated tissue of developing beans (Cotta et al. 2014). More precisely, transcripts of CaLTP3 (CaCc) homeolog were detected at different stages of pericarp development, while CaLTP1/2 (CaCe) homeologs were weakly expressed in this tissue. However, both CaLTP homeologs were highly expressed during the first stages of endosperm development. In another study, we also reported the high expression of CaCc and CaCe homeologs of CaLTP genes in the plagiotropic buds of the drought-tolerant cultivar “IAPAR59” subjected to water limitation but not in those of the drought-susceptible cultivar “Rubi” (Mofatto et al. 2016). This could be related to the thicker cuticle observed on the abaxial leaf surface in IAPAR59 compared to Rubi.
In a more recent study, Vieira et al. (2019) analyzed the expression of five FRIGIDA-like (FRL) genes in flowers, beans, and somatic embryos of C. arabica. As previously reported (Combes et al. 2013), gene silencing was not detected for CaFRL genes, both CaCc and CaCe homeologs being expressed in all tissues analyzed. However, HDE was observed, for example, during early stages of flower development with a bias toward the expression of CaCc homeolog of CaFRL2, while a bias toward a CaCc homeolog CaFRL4 was noticed in the latter stages of endosperm development. However, for this latter gene, a bias toward the overexpression of CaCe homeolog was observed in somatic embryos. This homeostasis of gene expression observed in the allopolyploid C. arabica could explain why this species had a greater phenotypic plasticity compared to its C. canephora parent (Bardil et al. 2011; Bertrand et al. 2015).
3 Gene Expression in Coffee Tissues
3.1 Beans
Several thousands of bean cDNAs were generated in the frame of the first coffee EST sequencing projects. For example, the Nestlé and Cornell project used three fruit libraries of C. canephora realized at early (whole cherries, 18–22 WAP), middle (endosperm and perisperm, 30 WAP [weeks after pollination]), and late (endosperm and perisperm, 42–46 WAP) stages of fruit development, leading to 9,843; 10,077; and 9,096 ESTs, respectively (Lin et al. 2005). On the other hand, the IRD and CENICAFE sequencing projects also generated, respectively, more than 5,800 ESTs from C. canephora and 9,500 ESTs from C. arabica but without mentioning the fruit developmental stage (Poncet et al. 2006; Montoya et al. 2007), while the BCGP project produced 14,779 ESTs from 2 fruit libraries (FR1 and FR2) of C. arabica and 15,162 from 2 libraries (FR4 and FV2) of C. racemosa (Vieira et al. 2006; Mondego et al. 2011).
Regarding the 700 genes reported in Tables 1, 2, and 3, most expression studies were performed in developing coffee beans in which it is not a surprise if we consider that the analysis of its transcriptome is absolutely required to understand the basis of genetic and environmental variations in coffee quality. The time between anthesis and full ripening varies between C. arabica (from 6 to 8 months) and C. canephora (from 9 to 11 months), and it is usually referred to as days (or weeks) after anthesis (DAA), flowering (DAF), or pollination (DAP) (De Castro and Marraccini 2006). The different stages of developing coffee cherries are mainly defined on its size and also in accordance to the changes of exocarp (pulp) color occurring during the latest maturation steps (Pezzopane et al. 2003; Morais et al. 2008; Gaspari-Pezzopane et al. 2012; Vieira et al. 2019).
Considering the bean and its own tissues, it is now very well known that some important changes occur during its development. Soon after fecundation and up to mid-development (e.g., 90–120 DAF for C. arabica), the bean is mainly constituted of perisperm (maternal) which is thereafter progressively replaced by the endosperm which hardens as it ripens during the maturation phase (Fig. 1). For a practical point of view, most of the gene expression studies performed during bean development (referred to as BD in Tables 1, 2, and 3) analyzed the bean as a whole without extracting RNA from separated perisperm and endosperm issues. If it is true to consider that perisperm represents the main tissue in the earliest stages of development (up to 90 DAF), this is no more the case after, when it is reduced to the fine silver skin membrane surrounding the bean. Several works analyzed expression in separated perisperm and endosperm tissues like those studying expansins and HMGRs (human 3-hydroxy-3-methyglutaryl-CoA reductase) (Budzinski et al. 2010) or enzymes of the mevalonic acid (MVA) pathway involved in the biosynthesis of cafestol and kahweol diterpenes (Tiski et al. 2011).

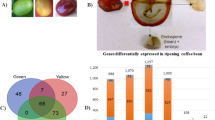
Schematic representation of the seven developmental stages and tissue changes occurring during fruit development of C. arabica. The time is indicated in days after flowering (DAF). Tissues: Pe perisperm, En endosperm, Pc pericarp. RT-qPCR gene expression profiles of CaCSP1, CaOLE-1, and CaManS1 (coding for 11S globulin, oleosin, and mannan synthase, respectively) are chosen to illustrate accumulation of storage proteins, triacylglycerols, and cell wall polysaccharides. Adapted from Dussert et al. (2018)
In 2008, Salmona et al. performed a transcriptomic approach combining targeted cDNA arrays, containing 266 selected candidate gene sequences and RT-qPCR on a large subset of 111 genes to decipher the transcriptional networks during the C. arabica bean development. This study was the first dividing coffee bean development in seven stages (ST1 0–60 DAF, small fruit with aqueous perisperm; ST2 60–90 DAF, perisperm surrounding a very small liquid endosperm; ST3 90–120 DAF, aqueous endosperm growing and replacing the perisperm; ST4 120–150 DAF, soft milky endosperm; ST5 150–210 DAF, hard white endosperm with green pericarp; ST6 210–240 DAF, ripening cherries with pericarp turning to yellow; ST7 > 240 DAF, mature cherries with red pericarp) (Fig. 1). Few years later, the same research group completed this study by combining gene expression and metabolite profiles (analyzed by high-performance liquid chromatography) in order to identify the key metabolic pathways of coffee bean development (Joët et al. 2009, 2010, 2012).
Regarding sucrose metabolism, Geromel et al. (2006, 2008b) reported high expression of CaSUS1, coding the sucrose synthase isoform 1, at the earlier stages of endosperm development (ST4), and high expression of CaSUS2 (sucrose synthase isoform 2) at the later stages of endosperm development (ST6–7) but also in the perisperm at 205 DAF (Joët et al. 2009). Even restricted at a fine membrane surrounding the endosperm, the high SUS2 expression detected at that time in the perisperm could contribute to the peak of sucrose detected at the latest development stages in both pericarp and endosperm tissues (Rogers et al. 1999b).
Together with other studies, the genes involved in the most important biochemical pathways were now studied like those involved in sucrose (Geromel et al. 2006, 2008b; Privat et al. 2008; Joët et al. 2014), raffinose (dos Santos et al. 2011, 2015; Ivamoto et al. 2017a) metabolism, polysaccharide synthesis such as galactomannans (Marraccini et al. 2005; Pré et al. 2008; Joët et al. 2014; Dussert et al. 2018), lipid synthesis and transport (Simkin et al. 2006; Cotta et al. 2014; Dussert et al. 2018), caffeine (Ogawa et al. 2001; Uefuji et al. 2003; Mizuno et al. 2003a, b; Koshiro et al. 2006; Perrois et al. 2015; Maluf et al. 2009; Kumar and Giridhar 2015; Kumar et al. 2017), chlorogenic acids (CGAs) (Lepelley et al. 2007, 2012b), carotenoids (Simkin et al. 2010), trigonellines (Mizuno et al. 2014), storage proteins (Marraccini et al. 1999; Simkin et al. 2006; Dussert et al. 2018), and dehydrins and LEAs (Hinniger et al. 2006) (Table 1). Altogether, these studies revealed the existence of several phases during coffee bean development. The first one (perisperm-specific) is characterized by the synthesis of CGA occurring early in the perisperm and accumulation of chitinases, as also confirmed by 2D gel electrophoresis and protein sequencing (De Castro and Marraccini 2006; Alves et al. 2016). More recently, Ivamoto et al. (2017a) performed the first large-scale transcriptome analysis of C. arabica beans during initial (from 30 to 150 DAF) developmental stages, showing the predominant expression of genes of catalytic protein, kinases, cytochrome P450, and binding site domains in the perisperm, for example. The second phase (between ST3 and ST6) is characterized by the activation of cell wall polysaccharide (mainly galactomannans and arabinogalactans) biosynthetic machinery and the synthesis of storage proteins (Marraccini et al. 1999; Pré et al. 2008; Joët et al. 2014; Dussert et al. 2018) (Fig. 1). The third phase concerns the metabolic rerouting of CGA characterized by the HCT1 expression peak during the latest stages of seed development and the synthesis, storage, and exports of fatty acids requiring oleosins and LTPs (lipid transfer proteins). Finally, the last (endosperm-specific) stage is characterized by the sucrose synthesis and accumulation and dehydration of beans. These steps were recently confirmed by the recent long-read sequencing full-length (LRS) coffee bean transcriptome (Cheng et al. 2018). In that case, the last steps of coffee bean development were characterized by the drastic drop of chitinase transcripts and the great upregulation of genes coding late embryogenesis abundant (LEA) proteins, heat-shock proteins (HSPs), and ROS (reactive oxygen species) scavenging (e.g., superoxide dismutases, catalases, glutathione reductases, glutaredoxins, and glutathione peroxidases) and antioxidant (e.g., dehydroascorbate reductases, glutathione reductases, monodehydroascorbate reductases, and thioredoxins) enzymes, for example (Dussert et al. 2018).
The regulation of gene expression during coffee bean development should implicate specific transcription factors (TFs). In a recent study, Dong et al. (2019a) identified 63 NAC-like genes in the reference genome of C. canephora, coding TFs well-known to play important functions in plant development and stress regulations (Puranik et al. 2012). After FPKM (Fragments Per Kilobase of transcript per Million mapped reads) treatment of RNAseq data generated at different stages of fruit development, these authors identified 54 CcNAC genes with DEG (differentially expressed gene) profiles during the bean development which were verified by qPCR for 10 of them. This led to classify the CcNAC genes with continuous upregulated expression as positive regulator of bean development, while those showing downregulated expression were considered as negatively correlated with bean development.
In addition to the gene expression studies performed during coffee bean development, several works also analyzed gene expression in beans during drying (Bytof et al. 2007; Kramer et al. 2010; Santos et al. 2013; Selmar et al. 2006) and germination (da Silva et al. 2019; Lepelley et al. 2012a; Marraccini et al. 2001; Santos et al. 2013) processes.
3.2 Leaves
In the frame of the Nestlé/Cornell (Lin et al. 2005) and IRD (Poncet et al. 2006) sequencing projects, 8,942 and 4,606 ESTs were generated from C. canephora leaves, respectively, while 12,024 ESTs were also sequenced from C. arabica leaves by CENICAFE (Montoya et al. 2007). On the other hand, the BCGP produced 26,931 ESTs from 4 leaf libraries (LV4, LV5, young leaves from orthotropic branches, and LV8, LV9, mature leaves from plagiotropic branches) of C. arabica, as well as 5,567 ESTs of C. arabica leaves infected with leaf miner and leaf rust (RM1 library), and 13,111 ESTs from 2 leaf libraries (SH1 and SH3) of C. canephora plants grown under water deficit (Vieira et al. 2006; Mondego et al. 2011; Vinecky et al. 2012). In this project, leaf ESTs were also generated in the SS1 (960 ESTs), SH2 (7,368 ESTs), and AR1-LP1 (5,664 ESTs) cDNA libraries from tissue pools of C. arabica plantlets well-watered, drought-stressed, and treated with arachidonic acid, respectively. Since these studies, numerous projects aiming to study the effects of biotic and abiotic stresses in leaves by RNAseq were performed (see Sects. 4 and 5).
In coffee, leaves are important organs not only as source organs performing photosynthesis and sugar biosynthesis (Campa et al. 2004) but also because they synthesize many other biochemical compounds such as caffeine (Frischknecht et al. 1986; Ashihara et al. 1996; Zheng and Ashihara 2004; Ashihara 2006), chlorogenic acids (CGAs) (Ky et al. 2001; Bertrand et al. 2003; Campa et al. 2017), and trigonelline (Zheng et al. 2004; Zheng and Ashihara 2004) which are further exported to beans and involved in the final cup quality (Leroy et al. 2006).
From the data of Tables 1, 2, and 3, leaf expression studies were reported for more than 400 genes. The first published concerned the three methyltransferases of the caffeine pathway encoded by the XMT (xanthosine N-methyltransferase), MXMT (7-methylxanthine-N-methyltransferase or theobromine synthase), and DXMT (3,7-dimethylxanthine-N-methyltranferase or caffeine synthase) genes (Ogawa et al. 2001; Uefuji et al. 2003; Mizuno et al. 2003a, b). These studies, initially performed by semiquantitative PCR, were further completed by RT-qPCR to better specify the expression of CaXMT1, CaMXMT1, and CaDXMT2 genes (belonging to the C. canephora sub-genome) and CaXMT2, CaMXMT2, and CaDXMT1 (belonging to the C. eugenioides sub-genome) in young and mature leaves of C. arabica and C. canephora (Perrois et al. 2015).
Numerous other studies also detailed the leaf expression profiles of genes of photosynthesis (Marraccini et al. 2003, 2011), sugar metabolism (Privat et al. 2008), and the biosynthetic pathways of carotenoids (Simkin et al. 2008), trigonelline (Mizuno et al. 2014), CGAs (Lepelley et al. 2007, 2012b), and diterpenes (Ivamoto et al. 2017b), for example.
3.3 Roots
More than 12,000 root ESTs were produced in the frame of the BCGP from 4 libraries (RT3, roots; NS1, root infected by nematodes; RT5, roots treated with acibenzolar-S-methyl – a systemic acquired resistance [SAR] inducer; and RT8, roots stressed with aluminum) of C. arabica (Vieira et al. 2006; Mondego et al. 2011). In 2006, 1,587 ESTs were produced from embryonic roots of two C. arabica cultivars (De Nardi et al. 2006). Among them, 1,506 sequences were used to set up a cDNA microarray which led to the identification of 139 genes differentially expressed in response to induced SAR. In the frame of PhD thesis of T.S. Costa (2014), 25,574 cDNA sequences were generated from roots of drought-susceptible and drought-tolerant clones of C. canephora Conilon submitted to water limitation. Even though these data were not deposited in public databases, this study permitted to identify several genes with upregulated expression under drought (see Sect. 5.1). In a more recent RNAseq study, dos Santos et al. (2019) obtained 34,654 assembled contigs from N-starved roots of C. arabica and identified three AMT (coding specific transporters of ammonium) and three NRT (coding nitrate transporters) for which in silico gene expression profiles (dos Santos et al. 2017) were validated by RT-qPCR (dos Santos et al. 2019). Expression profiles in roots were also reported for genes of sugar (Geromel et al. 2006) and caffeine (Ogawa et al. 2001) biosynthetic pathways.
3.4 Flowers
Compared to fruits, leaves, and roots, the studies analyzing gene expression in flowers are very limited. In terms of genetic resources, the BCGP generated 23,036 ESTs from 3 cDNA libraries (FB1, FB2, and FB4) of flowers in different developmental stages and 14,779 ESTs from 2 libraries (FR1 and FR2) corresponding to a mixture of transcripts extracted from flower buds and fruits at different developmental stages (Vieira et al. 2006; Mondego et al. 2011). The CENICAFE research group also reported the production of 8,707 EST sequences from flowers of C. arabica (cv. Caturra), but these data were neither released in public databases. In a recent RNAseq study, Ivamoto et al. (2017a) identified several genes that were exclusively expressed in flowers such as those coding a FASCICLIN-like arabinogalactan protein precursor (FLA3, a protein with InterPro FAS1 Domain IPR000782) and a pectin esterase inhibitor (InterPro Domain IPR006501).
The studies of Asquini et al. (2011) and Nowak et al. (2011), aiming to characterize S-RNase genes and to analyze their expression in pistils (at pre- and post-anthesis stages) and stamens of C. arabica and C. canephora flowers, were also worth noting.
Other studies characterized the genes of C. arabica coding MADS-box TFs (involved in the floral organ identity) and also checked the expression of FLOWERING LOCUS C (FLC), AGAMOUS, APETALA3, and SEPALLATA3 (de Oliveira et al. 2010, 2014). In a more recent study, Vieira et al. (2019) analyzed the expression of five FRIGIDA-like (FRL) genes, coding key proteins that regulate flowering by activating FLC (Wang et al. 2006). In that case, these authors used the qPCR TaqMAMA-based method (Li et al. 2004) to identify the expression of CaCc and CaCe homeologs of FRL genes in C. arabica flowers at different development stages (see also Sects. 2.3 and 3.5). Altogether, these results should help us to understand the genetic determinisms controlling the gametophytic self-incompatibility system of C. canephora (Berthaud 1980; Lashermes et al. 1996; Moraes et al. 2018) and coffee male sterility (Mazzafera et al. 1990; Toniutti et al. 2019a).
3.5 Somatic Embryogenesis
In coffee, the somatic embryogenesis (SE) is important particularly to propagate elite clones of C. canephora and F1 hybrids of C. arabica that could not be spread by seeds (Etienne et al. 2018; Bertrand et al. 2019; Georget et al. 2019). This is the reason why several laboratories are working to identify the genes controlling the main phases and key developmental switches of coffee SE. This also explains the important number (12) of cDNA libraries from suspension cells, calli (primary, embryogenic, and non-embryogenic), and embryos performed in the frame of the BCGP, which generated more than 65,000 ESTs (Vieira et al. 2006; Mondego et al. 2011).
Among these genes, it was reported that the expression of CcLEC1 (LEAFY COTYLEDON 1, a key regulator for embryogenesis) and CcBBM1 (BABY BOOM 1, a AP2/ERF TF associated with cell proliferation) was only observed after SE induction in C. canephora, whereas CcWOX4 (WUSCHEL-RELATED HOMEOBOX4, a plant regulator of embryogenic patterning and stem cell maintenance) expression decreased during embryo maturation (Nic-Can et al. 2013). The expression of BBM and SERK1 (somatic embryogenesis receptor-like kinase 1, a positive regulator of SE activating the YUCCA [flavin-containing monooxygenase]-dependent auxin biosynthesis) genes could also constitute a good parameter for evaluating the development and quality of C. arabica (Silva et al. 2014, 2015; Torres et al. 2015) and C. canephora (Pérez-Pascual et al. 2018) embryogenic cell suspensions. The fact that expression of FLC and FRL (especially that of CaFRL-3, CaFRL-4, and CaFRL-5) genes, initially reported as regulators of flowering development, was also observed in both zygotic and somatic embryos of C. arabica (Vieira et al. 2019) clearly indicates that both embryogenesis processes share common developmental pathways.
In order to better understand the transcriptomic changes occurring during SE process, Quintana-Escobar et al. (2019) recently performed the first RNAseq study analyzing different stages of SE induction in C. canephora. Among the genes differentially expressed, these authors identified eight ARF (auxin response factors) as well as seven Aux/IAA (auxin/indole-3-acetic acid regulators) and confirmed that CcARF18 and CcARF5 genes were highly expressed after 21 days of the SE induction. In another recent study, Pinto et al. (2019) characterized 17 GH3 genes from C. canephora (encoding the Gretchen Hagen 3 already reported to be key proteins controlling somatic embryogenesis induction through auxin) and analyzed their expression profiles in cells with contrasting embryogenic potential in C. arabica, showing that CaGH3.15 was correlated with CaBBM, a C. arabica ortholog of a major somatic embryogenesis regulator (Silva et al. 2015). Altogether, these genes could be useful as markers to follow the SE stage converting somatic to embryogenic cells.
4 Coffee Gene Expression in Response to Biotic Stress
Recent modeling studies have delivered warnings on the threat of climate change (CC) by increasing attacks by pests and pathogens (Avelino et al. 2004, 2015; Ghini et al. 2008, 2011, 2015; Jaramillo et al. 2011; Kutywayo et al. 2013; Magrach and Ghazoul 2015). For both C. canephora and C. arabica, the main pests and diseases are (1) the leaf rust caused by the fungus H. vastatrix, (2) the leaf miner Leucoptera coffeella (Guérin-Mèneville), (3) the root attacks caused by nematodes, (4) the fruit damages caused by the borer Hypothenemus hampei, and (5) the coffee berry disease (CBD) caused by the hemibiotrophic fungus Colletotrichum kahawae which is a major constraint of C. arabica coffee production in Africa (van der Vossen and Walyaro 2009).
Regarding the coffee genetic diversity, most of C. canephora are resistant to coffee leaf rust (CLR), while “pure” (non-introgressed) C. arabica are susceptible. However, Catimor and Sarchimor cultivars of C. arabica introgressed with the HdT are considered as totally or partially resistant to CLR (Eskes and Leroy 2004). Natural resistances to coffee berry borer (CBB) and coffee leaf miner (CLM) are rather limited in both C. canephora and C. arabica species. However, natural resistance to the CLM can be found in several wild coffee diploid species, such as in C. racemosa (Guerreiro-Filho et al. 1999; Guerreiro-Filho 2006), and has been introgressed into C. arabica to generate new cultivars (e.g., Siriema) resistant to CLR (Matiello et al. 2015). Regarding nematodes, a large genetic diversity exists particularly in diploid species (e.g., C. canephora, C. liberica, and C. congensis) but less in C. arabica, regarding the variation in resistance particularly to the root-knot Meloidogyne spp. from high susceptibility to near immunity as it is the case of the clone 14 of C. canephora Conilon (Lima et al. 2014, 2015). Information about genetic resistance to coffee berry borer (CBB) is very limited for both C. arabica and C. canephora species. However, Romero and Cortina (2004, 2007) reported a reduction of CBB growth rate when H. hampei is fed with C. liberica fruits. In another study, Sera et al. (2010) showed that C. kapakata, Psilanthus bengalensis, C. eugenioides, as well as genotypes introgressed with C. eugenioides were CBB resistant. In that case, the CBBR of C. eugenioides and C. kapakata was observed at the pericarp level (but not in the bean), while P. bengalensis presented CBBR in both tissues. In addition to be CLRR, some C. arabica coming from HdT, but also the F1 hybrid cultivar Ruiru 11, were also reported as CBDR (Omondi et al. 2004, Walyaro 1983; Van der Vossen 1985). This genetic diversity observed in the Coffea genus regarding these different abiotic stresses could be used to identify the genes controlling these resistances and to initiate new breeding programs aiming to create new hybrids better resistant to pests and diseases.
On the other hand, the BCGP produced more than 5,000 ESTs of C. arabica from RM1 (leaves infected with CLM and CLR) and NS1 (roots infected with nematodes) (Vieira et al. 2006; Mondego et al. 2011. In a recent study, genes coding for the LOX (lipoxygenase), AOS (allene oxide synthase), AOC (allene oxide cyclase), and OPR (12-oxo-phytodienoic acid reductase) enzymes involved in the production of jasmonic acid (JA), one of the key plant hormones involved in plant defense against insect pests, were identified in C. canephora by bioinformatic approaches (Bharathi and Sreenath 2017) but without confirming gene expression of this pathway in infested coffee plants.
4.1 Coffee Leaf Rust (CLR)
In 2004, Fernandez et al. used suppression subtractive hybridization (SSH) method and semiquantitative RT-PCR to identify C. arabica L. genes involved in the specific hypersensitive reaction (HR) upon infection by H. vastatrix. Among the genes showing HR upregulation were those coding for receptor kinases, AP2 domain and WRKY TFs, cytochromes P450, heat-shock 70 proteins, several glucosyltransferases, and NDR1, for example. Other studies showed that SA and MeJA treatments markedly upregulated the expression of CaNDR1 (coding a non-race-specific disease resistance protein well-known to be involved in resistance signalization pathway in Arabidopsis thaliana) and CaWRKY1 genes, suggesting a key role of their corresponding proteins in the molecular resistance responses of coffee to H. vastatrix (Ganesh et al. 2006; Cacas et al. 2011; Petitot et al. 2008, 2013). This was confirmed by Ramiro et al. (2010) who showed that in addition to CaWRKY1, expression of CaWRKY3, CaWRKY17, CaWRKY19/20/21, and CaWRKY22 genes was also highly upregulated upon CLR. Although a significant correlation was also observed between WRKY expression profiles after MeJA and rust treatments, expression of coffee genes involved in JA biosynthesis, including allene oxide synthase (CaAOS) and lipoxygenase (Ca9-LOX and Ca13-LOX), did not support the involvement of JA in the early coffee resistance responses to CLR.
The first valuable EST dataset from C. arabica CIFC 147/1 (CLR resistant) infected by leaf rust was produced by Fernandez et al. (2012) who identified 205,089 ESTs and 13,951 contigs from coffee together with 57,332 ESTs and 6,763 contigs from H. vastatrix. Among the most abundant coffee genes expressed in rust-infected leaves were those coding for several pathogenesis-related (thaumatin-like) proteins and enzymes of carbohydrate, amino acid, and lipid transport/metabolism. Florez et al. (2017) also used the C. arabica cultivars Caturra (CLR susceptible) and HdT CIFC 832/1 (CLR resistant) to generate 43,159 contigs which were assembled using as a reference the genome of C. canephora (Denoeud et al. 2014). Among DEG profiles identified by RT-qPCR were genes coding for a putative disease resistance protein RGA1, putative disease resistance response (dirigent-like protein) family protein, and Premnaspiridione oxygenase with higher expression at early stage of rust infection in the resistant cultivar plant than in the susceptible genotype. In addition, expression of several TFs (putative basic helix-loop-helix bHLH DNA-binding superfamily protein and ethylene-responsive transcription factor 1B) was detected earlier in HdT than in Caturra, suggesting that they may be involved in the defense mechanisms of the CLRR cultivar. In a more recent study, Echeverría-Beirute et al. (2019) performed RNAseq approach to study the effects of CLR and fruit thinning in leaves of susceptible cultivars red Catuaí (Caturra x Mundo Novo) and F1 hybrid H3 (Caturra x Ethiopian 531) of C. arabica. Using regression and prediction statistical models, these authors identified 460 DEGs between the inbred and the F1 hybrid. Among them, the expression of PR (pathogenesis-related) genes was upregulated in Catuaí, while those coding proteins involved in homoeostasis increased in the F1 hybrid. Even though these results were not confirmed by RT-qPCR, they validate the hypothesis of lower impact of CLR in F1 hybrids (Echeverria-Beirute et al. 2018) due to their physiological status, which itself depends on their genetic background, plant vigor, agronomic conditions, and environmental factors (Toniutti et al. 2017, 2019b).
4.2 Coffee Leaf Miner (CLM)
Although the defense mechanisms to leaf miner are not well understood, previous genetic analyses suggested that this resistance was dominant and controlled by a limited number of genes (Guerreiro-Filho et al. 1999). The first attempt to identify these genes was performed by SSH method coupled with the screening of DNA macroarrays to study gene expression in the leaves of the CLM-susceptible (CLMS: red Catuaí) and CLM-resistant cultivar (CLMR corresponding to a backcross of [C. racemosa x C. arabica x C. arabica]) infested by L. coffeella (Mondego et al. 2005). From the 1,500 ESTs spotted on the array, upregulated expression upon CLM infestation was observed for several ESTs coding proteins previously reported to be related to plant defense and biotic stress and similar to the phospholipase D, the lipoxygenase LOX3, the late embryogenesis abundant protein 1 (LEA1), the acid phosphatase vegetative storage protein (VSP), and the lipid transfer protein/trypsin inhibitor/seed storage domain, for example. For CaPR8 (class III chitinase), CaSPC25 (signal peptidase complex subunit), CaPSAH (photosystem I), CaCAX9 (a putative calcium exchanger), and CaBEL (BEL1-related homeotic protein 29) genes, their upregulated expression upon CLM infestation suggested that they play a key role in coffee defense mechanisms against L. coffeella.
In a more recent study, Cardoso et al. (2014) used a 135 K microarray (NimbleGen) based on the 33,000 genes identified in the frame of the BCGP, to identify DEG genes in CLMS and CLMR cultivars of C. arabica at three stages (T0, non-infected/control; T1, egg hatching, and T2, egg eclosion) of interaction with L. coffeella. Even though previous studies reported that caffeine has no effect on leaf miner survival rates (Guerreiro-Filho and Mazzafera 2000; Magalhães et al. 2010), high upregulated expression of a putative caffeine synthase gene was reported at both T0 and T2 in CLMR leaves compared to CLMS ones. In the same study, expression profiles of genes involved in plant response pathways to herbivory attacks (e.g., linoleic acid cycle, phenylpropanoid synthesis, and apoptosis), as well as JA (e.g., coding lipoxygenase and enoyl-CoA hydratase) and flavonoids (e.g., coding chalcone synthase and flavanone 3-hydroxylase-like) biosynthesis, were also upregulated in CLMR plants even in the absence (at T0) of leaf miner infestation, indicating that defense was already built up in these plants prior to infection, as a priming mechanism.
4.3 Nematodes (NEM)
Despite the important damages caused by nematodes, there are a limited number of studies analyzing the coffee gene responses to these pathogens. When studying WRKY genes coding transcription factors regulating plant responses to biotic stresses, Ramiro et al. (2010) reported that expression of CaWRKY6, CaWRKY11, CaWRKY12, CaWRKY13/14, CaWRKY15, and CaWRKY17 genes was upregulated in roots of C. arabica cv. IAPAR59 infected by the RKN Meloidogyne exigua. In another work, Severino et al. (2012) reported upregulated expression of CaPRX (encoding a putative class III peroxidase) in roots inoculated with RKN M. paranaensis but with significant difference between susceptible (C. arabica cv. Catuaí) and resistant (C. canephora cv. Robusta) plants. The nematode-resistant (NEMR) clone 14 of C. canephora Conilon (Lima et al. 2014, 2015) was also used to investigate gene expression in roots at regular days after infestation (4, 8, 12, 20, 32, and 45 DAI) by the root-knot M. paranaensis (Lima 2015). The RNAseq data (not yet publicly available) showed higher expression levels of several PR (pathogenesis-related) genes, such as those coding class III chitinase and NBS-LRR proteins, in infected roots of NEMR clone 14 than in those of NEMS clone 22. In addition, the peak of NBS-LRR transcripts was detected at 8 and 20 DAI for the clones 14 and 22, respectively, suggesting earlier expression of this gene in NEMR than in NEMS coffee clones (Valeriano et al. 2019). RT-qPCR experiments also showed that expression of CcCPI1 (coding a cysteine proteinase inhibitor) was higher in roots of clone 14 than in those of 22, with or without nematode infestation, suggesting that the this protein, also highly expressed in coffee beans under development and germination (Lepelley et al. 2012a), could also play a key role in controlling nematode development. In that sense, CPIs have already been reported to inhibit proteinases in the digestive tracts, therefore reducing the destructive effects of herbivorous insects (Benchabane et al. 2010; Schluter et al. 2010), and to increase tolerance to nematodes as well as to fungal and bacterial pathogens in transgenic plants (Urwin et al. 2003; Martinez et al. 2005).
4.4 Coffee Berry Borer (CBB)
Considering that C. arabica fruits are more susceptible to CBB than those of C. liberica, Idárraga et al. (2012) constructed cDNA libraries from fruits for these two species infested with H. hampei and generated 3,634 singletons and 1,454 contigs. In silico analyses revealed that infested C. arabica berries displayed a higher number of DEG genes coding proteins involved in general stress responses, while genes coding proteins involved in insect defense were overexpressed in C. liberica. For some of these genes, expression profiles in infested cherries were checked by RT-qPCR. Interestingly, expression levels of genes coding a hevein-like protein, an isoprene synthase, a SA carboxyl methyltransferase, and a patatin-like protein appeared much more upregulated in C. liberica than in C. arabica. The upregulation of these genes was already reported in other plants in response to insect herbivory and JA treatments (Kiba et al. 2003; Reymond et al. 2000; Falco et al. 2001), suggesting that they could be involved in the partial resistance to CBB in C. liberica.
4.5 Coffee Berry Disease (CBD)
Cytological and biochemical studies revealed that coffee resistance to C. kahawae is characterized by restricted fungal growth associated with several host responses, such as hypersensitive-like cell death (HR), callose deposition, accumulation of phenolic compounds, lignification of host cell walls, and increased activity of oxidative and peroxidase enzymes (Silva et al. 2006; Gichuru 1997, 2007; Loureiro et al. 2012).
The first study analyzing gene expression in response to C. kahawae was performed by Figueiredo et al. (2013) in hypocotyls of C. arabica cultivars Catimor 88 (HdT derivative CBDR) and Caturra CIFC 19/1 (CBDS). These authors showed that expression levels of RLK (coding a receptor-like kinase) and PR10 (coding a pathogenesis-related protein 10) genes were higher in Catimor than in CBD-infected Caturra. Interestingly, upregulated expression of these two genes was also reported during coffee infection with H. vastatrix (Fernandez et al. 2004). In order to understand the molecular mechanisms involved in coffee resistance to C. kahawae, Diniz et al. (2017) evaluate the expression of genes involved in SA, JA, and ethylene (ET) pathways in the same cultivars. From the 14 genes studied by RT-qPCR, these authors showed the involvement of JA and ET phytohormones rather than SA in this pathosystem. Regarding the ET pathway, the strong activation of ERF1 gene (coding for ET receptor) at the beginning of the necrotrophic phase suggests the involvement of ethylene in tissue senescence.
4.6 Gene Expression in Response to Other Pests and Diseases
Of the two commercially cultivated coffee species, C. arabica and C. canephora are considered as susceptible and resistant, respectively, to the insect pest Xylotrechus quadripes known as coffee white stem borer (CWSB). Using SSH approach, Bharathi et al. (2017) identified 265 unigenes overexpressed in C. canephora bark tissues upon CWSB larval infestation, many of them coding putative pectin-degrading enzymes like a pectate lyase (Cc07_g00190Footnote 1), three polygalacturonases (Cc03_g15700, Cc03_g15740, and Cc03_g15840), and a pectinacetylesterase (Cc08_g04630). By RT-qPCR, these authors also showed that the expression of Cc07_g00190 was strongly induced at 72 h after CWSB infestation. The possible role of this pectinolytic enzyme in the production of oligogalacturonides was proposed, which could act as elicitors involved in defense responses of C. canephora to CWSB (Bharathi and Sreenath 2017).
5 Coffee Gene Expression in Response to Abiotic Stress
Several models predicted that CC will have strong negative impacts on both C. canephora and C. arabica species at environmental, economic, and social levels (Assad et al. 2004; Bunn et al. 2015a, b; Ovalle-Rivera et al. 2015; Davis et al. 2012, 2019; Moat et al. 2017, 2019). Drought and high air temperatures are undoubtedly the major threats to coffee production, forecasted by potential climate changes (IPCC 2013). Drought is a limiting factor that affects flowering and yield of coffee (DaMatta and Ramalho 2006), as well as bean development and biochemical composition and consequently the final cup quality (Silva et al. 2005; Vinecky et al. 2017). Increased [CO2] in air is also a key factor for coffee plant acclimation to high temperature; strengthening the photosynthetic pathway, metabolism, and antioxidant protection; and modifying gene transcription and mineral balance (Ramalho et al. 2013; Martins et al. 2014, 2016; Ghini et al. 2015; Rodrigues et al. 2016). In this context, understanding the genetic determinism of coffee’s adaptation to abiotic stress has become essential for creating new varieties (Cheserek and Gichimu 2012).
5.1 Drought
The first study analyzing the effects of drought stress was performed by Simkin et al. (2008), who reported the gene expression profiles of the carotenoid biosynthesis pathway in leaf, branch, and flower tissues of C. arabica subjected to water withdrawal. In this work, it was shown that the transcript levels of PTOX, CRTR-B, NCED3, CCD1, and FIB1 increased under drought, suggesting that drought favored the synthesis of xanthophylls implicated in the adaptation of plastids to changing environmental conditions by preventing photooxidative damage of the photosynthetic apparatus. On the other hand, drought was reported to decrease the RBCS1 gene expression in both C. arabica and C. canephora species (Marraccini et al. 2011, 2012). However, this reduction was not accompanied by a decrease of RBCS1 protein in the leaves of C. canephora under water withdrawal. In the same work, it was also shown that the transcriptional contribution of each RBCS1 homeolog may be affected by drought in C. arabica cultivars (Marraccini et al. 2011). In C. canephora, and whatever the clone studied, drought was also shown to downregulate the leaf expression of many genes related to photosynthesis such as CcCAB1 (coding chlorophyll a-/b-binding proteins), CcCA1 (coding for the carbonic anhydrase supplying CO2 for Rubisco), as well as expression of CcPSBO, CcPSBP, and CcPSBQ genes coding proteins of the PSII oxygen-evolving complex (Marraccini et al. 2012; Vieira et al. 2013).
On the other hand, drought stress significantly upregulated the expression of genes coding proteins involved in maintenance, reinforcement, and protection during the dehydration-rehydration process such as dehydrins and glycin-rich and heat-shock proteins in C. canephora (Marraccini et al. 2012; Vieira et al. 2013) and C. arabica (Santos and Mazzafera 2012; Mofatto et al. 2016). Drought stress was also shown to increase expression of some PIP (plasma membrane intrinsic proteins) genes in the leaves and roots of different coffee species, suggesting the involvement of these aquaporins in controlling the water status in coffee plants (dos Santos and Mazzafera 2013; Miniussi et al. 2015).
In coffee, like in many other plants, drought stress was also reported to affect the metabolic pathways involved in the synthesis of many solutes such as sugars of the raffinose family oligosaccharides (RFOs) (e.g., trehalose, raffinose, and stachyose), already described to be involved in osmoprotection against abiotic stresses in plants (Kerepesi and Galiba 2000). The upregulated expression of CaGolS2 and CaGolS3 genes coding galactinol synthases explained the increase of raffinose and stachyose contents also observed in leaves of C. arabica cv. IAPAR59 plants submitted to severe water deficit (dos Santos et al. 2011). In C. canephora Conilon, water limitation also increased CcGolS1 gene expression in leaves of the drought-tolerant (DT) clone 14 but decreased the expression of the same gene in leaves of the drought-susceptible (DS) clone 109A (dos Santos et al. 2015). Drought was also shown to upregulate the expression of M6PR gene coding the mannose-6-phosphate reductase in leaves of both C. canephora (Marraccini et al. 2012) and C. arabica (Freire et al. 2013). In C. arabica cv. IAPAR59, the increased expression of CaPMI (mannitol synthesis) and decreased CaMTD (controlling mannitol degradation) expression under drought were correlated with high mannitol levels detected in leaves under drought conditions (de Carvalho et al. 2014).
Drought also increased the expression of regulatory genes CcRD29, CcRD26, and CcDREB1D coding a RD29-like protein, a NAC-RD26-like TF, and an AP2/ERF DREB-like TF, respectively, in DT (14, 73, and 120) and DS (22) clones of C. canephora Conilon (Marraccini et al. 2012; Vieira et al. 2013). Even though these studies highlighted the existence of different mechanisms among the DT clones of C. canephora regarding water deficit, they also showed that CcDREB1D expression was always higher in leaves of DT clones (particularly in clone 14) than in those of DS clone 22 under water withdrawal (Fig. 2). Upregulated expression of the CcDREB1D was also reported in leaves of C. canephora and C. arabica subjected to low relative humidity (Thioune et al. 2017; Alves et al. 2018). A study of CcDREB1D promoter regions in the DT clone 14 and DS clone 22 revealed the existence of several haplotypes diverging by several SNPs and insertions/deletions (Alves et al. 2017). A functional analysis of these promoters in transgenic plants of C. arabica var. Caturra showed that haplotype HP16 (found in the DT clone 14) was able to drive the expression of the uidA reporter gene under water deficit in leaf mesophyll and guard cells more strongly and earlier than the HP15 (present in both clones) and HP17 (only present in DS clone 22) haplotypes (Alves et al. 2017). In a more recent work aiming to study the expression of DREB-like genes regarding various abiotic stresses (Torres et al. 2019), drought (mimicked by water limitation) was shown to upregulate expression of CcDREB1B, CcRAP2.4, CcERF027, CcDREB1D, and CcTINY mainly in leaves of C. canephora DT clones, while drought (mimicked by low humidity) upregulated the expression of CaERF053, CaRAP2.4, CaERF017, CaERF027, CaDREB1D, and CaDREB2A.1 in leaves of C. arabica. On the other hand, expression of CcDREB2F, CcERF016, and CcRAP2.4 genes was greatly upregulated under drought specifically in the roots of DS clone 22 (Fig. 2), which could help this clone to compensate its low efficiency in controlling stomatal closure and high reduction of net CO2 assimilation (A) observed upon drought acclimation (Marraccini et al. 2012).

Gene expression profiles of DREB-like genes in leaves and roots of DT (14, 73, and 120) and DS (22) clones of C. canephora Conilon subjected (NI not irrigated, black isobars) or not (I irrigated, white isobars) to water limitation. The DT and DS clones are separated by a vertically dotted line. Gene names are indicated in the histograms. Expression values corresponding to the mean of three biological and technical replications (±SD) are expressed in fold change relative to the expression level of the sample 22I as the reference sample (relative expression = 1). Transcript abundances were normalized using the expression of the CcUBQ10 (Barsalobres-Cavallari et al. 2009) as the endogenous control. Treatments sharing the same letter are not significantly different. Data adapted from Torres et al. (2019)
M.G. Cotta (2017) also analyzed the expression profiles of genes coding the PYR/PYL/RCAR-SnRK2-PP2C proteins known to be involved in the first steps of ABA perception and signal transduction in plants (Klingler et al. 2010), in leaves, and in roots of DT (14, 73, and 120) and DS (22) clones of C. canephora subjected to drought. In leaves, drought downregulated the expression of CcPYR1, CcPYL2, and CcPYL4 genes (coding ABA receptors) and upregulated the expression of CcAHG2 and CcHAB (coding PP2C phosphatases functioning as negative regulators of ABA pathway) in DT clones. However, expression of SnRK2 genes (coding protein kinases functioning as positive regulators of this pathway) was poorly affected by drought conditions. On the other hand, drought upregulated the expression of PP2C (e.g., CcABI1, CcABI2, CcAHG3) and SnRK2 (e.g., SnRK2.2, SnRK2.6, and SnRK2.7) genes mainly in roots of C. canephora DT clone 120. CcPYL8b was the gene most expressed in drought-stressed roots, particularly in DT clones 73 and 120, while expression of CaPYL8a was upregulated by drought mainly in leaves of C. arabica DT accession (Santos et al. 2019).
In C. canephora, Menezes-Silva et al. (2017) reported that coffee plants exposed to multiple drought events tended to display a higher expression of the RD29B and RD22 genes which could be involved in acclimation to repeated drought events. Recently, de Freitas Guedes et al. (2018) performed an RNAseq study to analyze the effects of multiple drought stress on gene expression in leaves of the DT clone 120 and DS clone 109 of C. canephora. Among the 22,764 genes generated, these authors identified 49 genes in the DT clone (e.g., coding a MYB-like proteins or for defense-related proteins containing LRR and kinase domains), which could be involved in stress “memory.”
As previously mentioned, Costa (2014) analyzed the expression profiles of several genes in roots of DS and DT clones of C. canephora Conilon submitted to water limitation. Among the identified DEGs, it is worth noting that upregulated expression was specifically observed under drought in roots of the DT clone 14 for the CcMJE1 (coding a protein involved in MeJA metabolism), CcNCED3 (encoding a rate-limiting protein involved in the synthesis of abscisic acid), CcPAP1 (coding a putative protein containing the acid phosphatase domain TIGR01675 characterizing vegetative storage proteins (VSPs)), CcPRX1 (coding for a putative peroxidase), and CcclXIP (coding a chitinase-like xylanase inhibitor protein), as well as CcM6PR, CcGOLS3b, and CcLTP4 (involved in RFOs and lipid biosynthesis pathways) genes. More recently, Vasconcelos et al. (2011) reported that the protein expressed from the CaclXIP cDNA (originally identified as a class III chitinase encoding gene from C. arabica) functioned as a chitinase-like xylanase inhibitor protein (clXIP) of fungal xylanases. Altogether, these responses suggest the existence of cross talk between abiotic and biotic pathways in roots of DT clone 14 which could explain its drought tolerance and resistance to several species of RKN of Meloidogyne spp. (see Sect. 4.3).
It is also worth noting that expression of many genes cited in this section (e.g., coding dehydrins, enzymes of carotenoid and RFO pathways, and other proteins involved in stabilization of membranes and proteins) was also studied during the last stages of coffee bean development (Hinniger et al. 2006; Simkin et al. 2010; Ivamoto et al. 2017a; Dussert et al. 2018), characterized by the intense dehydration of endosperm (De Castro and Marraccini 2006; Eira et al. 2006).
5.2 High Temperature
The study of Bardil et al. (2011) was the first to analyze the effects of low (LT, day 26°C/night 22°C) and high (HT, day 30°C/night 26°C) temperature on homeologous genes expressed in leaves of C. arabica and in those of its two ancestral parents, C. canephora and C. eugenioides. Among the 15 K unigenes analyzed, around 50% appeared differentially expressed (with 25% upregulated) at low temperature between C. arabica, C. canephora, and C. eugenioides. Similar proportions were found at high temperature when comparing the transcriptome of C. arabica vs. C. eugenioides and C. canephora vs. C. eugenioides. However, only 8.9% of transcriptome divergence was observed when comparing C. arabica vs. C. canephora. In terms of expression patterns observed in C. arabica, the number of genes with “C. canephora-like dominance” increased from 8–14% under LT (in the Java and T18141 cultivars) to 21–26% under HT conditions. In that case, it was worth noting that transcription profiles of T18141 (a cultivar recently introgressed with C. canephora genome) were more similar to that of C. canephora than that of the “pure” (non-introgressed recently) Java cultivar. Altogether, these results indicate that C. arabica mainly expressed genes from its CaCc sub-genome under hot temperatures.
In another work, Bertrand et al. (2015) analyzed gene expression profiles in leaves of C. arabica, C. eugenioides, and C. canephora (cv. Nemaya) exposed to four thermal regimes (TRs: 18–14, 23–19, 28–24, and 33–29°C). Under hot temperatures, upregulated expression in C. arabica was observed for several genes like Cc10_g00570 coding a catalase (CAT3) (when compared to C. canephora) and Cc06_g11950 coding a photosystem II subunit X (when compared to C. eugenioides). On the other hand, expression profiles of Cc05_g04680 coding a L-ascorbate oxidase homolog and those of photosynthetic genes coding light-harvesting complex (LHCII: Cc04_g16410) and chlorophyll a–b-binding protein (CAB: Cc10_g00140, Cc05_g12720, Cc09_g09020, Cc05_g09650, and Cc09_g09030), or for respiration-like genes Cc10_g00410, Cc02_g25840, and Cc07_g00550 (coding a chloroplast glyceraldehyde-3-phosphate dehydrogenase, a chloroplast ribose-phosphate pyrophosphokinase, and a Rubisco methyltransferase, respectively), were strongly downregulated in C. arabica compared with its two parents.
In leaves of C. arabica, heat-shock conditions also upregulated the expression of CaGolS1, CaGolS2, CaGolS3, CaPMI, CaMTD, and CaERF014 and downregulated expression of CaM6PR (dos Santos et al. 2011; de Carvalho et al. 2014; Torres et al. 2019). The interactions of high temperature and high [CO2] on expression profiles of gene coding protective and antioxidant proteins were also studied by Martins et al. (2016) and Scotti-Campos et al. (2019) (see Sect. 5.4 below).
5.3 Cold Stress
The first studies to analyze the effects of cold stress on coffee gene expression were realized by Fortunato et al. (2010) and Batista-Santos et al. (2011) who subjected several cultivars and hybrids of C. canephora, C. arabica, and C. dewevrei to gradual cold treatments. These authors showed that upregulation of CaGRed and CaDHAR genes (coding a glutathione reductase (GR) and dehydroascorbate reductase, respectively) and of CaCP22, CaPI, and CaCytf (coding proteins involved in PSII, PSI, and Cytb6/f complex, respectively) could explain the ability of Icatu (C. arabica × C. canephora) cultivar to better support cold stress by reinforcing its antioxidative capabilities and maintaining efficient thylakoid functioning.
In their analysis of gene expression profiles in leaves of C. arabica, C. eugenioides, and C. canephora (cv. Nemaya) exposed to different thermal regimes, Bertrand et al. (2015) also reported upregulated expression profiles under cold stress in C. arabica for Cc07_g15610 gene coding a L-ascorbate oxidase, for genes involved in respiration (e.g., Cc02_g08980, Cc00_g15710, and Cc02_g06960 coding a phosphoenolpyruvate carboxylase kinase, a ribulose bisphosphate carboxylase small chain, and a sedoheptulose-1,7-bisphosphatase, respectively), and also for genes of photosynthesis (e.g., Cc02_g28520, Cc05_g15930, Cc07_g10820, and Cc06_g19130 coding a ferredoxin-nitrite reductase, a photosystem II 10 kDa polypeptide, a ferredoxin-NADP reductase, and a ferredoxin-dependent glutamate synthase, respectively). On the other hand, overexpression of Cc05_g10250, Cc00_g35890, and Cc05_g10310 genes (coding polyphenol oxidases) was seen in C. canephora under low temperatures. For the LHY (late elongated hypocotyl, Cc02_g39990) gene involved in circadian cycle, RT-qPCR experiments confirmed in silico data, showing the highest expression under low than high temperatures particularly in leaves of C. canephora.
More recently, two studies investigated the effects of cold stress on the leaf gene expression in C. canephora. In the first one, Dong et al. (2019a) performed gene expression analyses in leaves of C. canephora plants subjected to cold stress (C1 (7 days at day 13°C/night 8°C) followed by C2 (3 days at day 4°C/night 4°C)) but also in fruits at different stages of development. For the 38 CcNAC genes analyzed by qPCR in cold-stressed leaves, expression was (1) upregulated upon C1 and C2 treatments for 4 genes, (2) downregulated upon C1 (but not C2) for 10 genes, (3) upregulated upon C2 (but not C1) for 7 genes, and (4) downregulated upon both cold treatments for 17 genes. In the second work, the same authors characterized 49 CcWRKY genes from the reference genome of C. canephora and analyzed their expression profiles by qPCR for 45 of them in cold-stressed leaves as reported in the previous study (Dong et al. 2019b). This led to identify 14 CcWRKY genes with expression induced during the cold acclimation stage (upon C1 and C2 treatments), 17 genes upregulated by cold treatment (C2 but not C1), and 12 downregulated by both cold stress treatments. Among the 14,513 putative target genes of CcWRKY identified in C. canephora by a genome-wide analysis, 235 were categorized into response to the cold process, including carbohydrate metabolic, lipid metabolic, and photosynthesis process-related genes. Like in many other plants, these observations clearly highlight the vital regulatory role played by WRKY TFs in various developmental and physiological processes (such as seed development) but also in a range of abiotic stress (like cold, heat, drought, as well as salinity) and biotic stress (Rushton et al. 2010).
In a more recent work, Ramalho et al. (2018a) analyzed the impacts of single and combined exposure to drought and cold stress in C. arabica cv. Icatu, C. canephora cv. Apoatã, and the hybrid C. arabica cv. Obatã. At the physiological level, the Icatu cultivar showed a lower impact upon exposure to cold and drought stress, characterized by a reduced lipoperoxidation under stress interaction, for example. At the molecular level, simultaneous exposure of Icatu to both stresses increases the expression of genes coding ascorbate peroxidase (APX) involved in H2O2 removal (e.g., APXc [cytosolic] and APXt+s [stromatic]) and consequently total APX enzymatic activity. To a lesser extent, this situation was also observed in C. canephora, while Obatã was the less responsive genotype considering the studied genes.
5.4 CO2 Concentration
The research group of J.C. Ramalho (Lisbon University, Portugal) published several articles studying the effects of elevated [CO2] on coffee. They demonstrated that elevated [CO2] mitigated the impact of heat on coffee physiology (Rodrigues et al. 2016) and also contributed to preserve the bean quality (Ramalho et al. 2018b). In a study aiming to analyze the interactions of elevated [CO2] and high temperature on protective response mechanisms in coffee, Martins et al. (2016) showed that the maintenance (or increase) of the pools of several protective molecules (e.g., neoxanthin, lutein, carotenes, α-tocopherol, heat-shock proteins HSP70, and raffinose), activities of antioxidant enzymes (e.g., superoxide dismutase, APX, GR, and catalase [CAT]), and the upregulated expression of ELIP (coding chloroplast early light-induced protein) and Chap20 (coding chloroplast 20 kDa chaperonin) genes were correlated with heat tolerance (up to day 37°C/night 30°C) at 380 and 700 μL CO2 L−1 for both C. arabica L. cvs. Icatu and IPR108 and C. canephora cv. Conilon clone 153. These authors also showed that upregulated expression of genes related to protective (ELIPS, HSP70, Chap20, and Chap60) and antioxidant (CAT, APXc, APXt+s) proteins was largely driven by temperature, while enhanced [CO2] promoted a greater upregulation of these genes mainly in C. canephora CL153 and C. arabica Icatu. In the more recent study analyzing the expression of genes related to lipid metabolism under elevated [CO2], heat, and their interaction, Scotti-Campos et al. (2019) showed that the strong remodeling (unsaturation degree) of membrane lipids observed during the heat shock (from day 37°C/night 30°C to day 42°C/night 34°C) of plants grown under high [CO2], coordinated with FAD3 (coding for fatty acid desaturase) downregulation in C. arabica and upregulation of lipoxygenase-coding genes LOX5A (in CL153 and Icatu) and LOX5B (in Icatu), could contribute to long-term acclimation of coffee chloroplast membranes to climate changes.
5.5 Salt Stress
In leaves of C. arabica cv. IAPAR59, upregulated expression of galactinol synthase genes CaGolS2 and CaGolS3 was observed after irrigation with 150 mM NaCl (dos Santos et al. 2011). In the same cultivar, salt stress upregulated the expression of CaM6PR and CaPMI genes and markedly downregulated that of CaMTD (de Carvalho et al. 2014). In parallel, leaf mannitol contents increased gradually to reach a peak after 12 days of salt stress imposition. However, this content was lower than in leaves of plants under water deprivation, indicating that coffee plants have different responses to drought and salinity.
The effects of salt stress in leaves were recently studied by RNAseq in leaves of C. arabica seedlings irrigated with normal water (control, ECw [electrical conductivity] = 0.2 dS.m−1) or with deep sea water (salt treatment, ECw = 2.3 dS.m−1) (Haile and Kang 2018). From the 19,581 genes aligned on the reference genome of C. canephora, in silico analyses identified 611 genes presenting significant DEG profiles between the control and salt treatment. Among the most expressed upregulated genes were Cc00_g13890, Cc04_g05080, and Cc08_g11060, coding for WRKY TFs; Cc06_g01240 coding a putative trihelix TF GT-3a already reported in controlling the developmental process and response to abiotic and biotic stress (Park et al. 2004; Wang et al. 2016); and Cc10_g04710 coding the putative ethylene-responsive (ERF011) TF. On the other hand, salt stress also downregulated the expression of Cc05_g16570 (coding a putative MYB family transcription factor APL), Cc02_g17440 and Cc07_g03240 (both coding putative bHLH TFs), and Cc02_g10740 and Cc06_g21410 (coding putative transcription elongation factor SPT of RNA polymerase II). However, the DEG expression profiles of these TF-encoding genes were not verified in vivo by qPCR experiments.
5.6 Wounding
WRKY and NDR genes were previously reported as playing key roles in the molecular resistance responses of coffee to H. vastatrix (see Sects. 2.3 and 4.1). In the first study, Ganesh et al. (2006) reported upregulated expression of CaNDR1, CaWRKY1 (see Sect. 4.1), and CaR111 (coding a putative protein of unknown function) genes in leaves of C. arabica wounded by performing transversal cuts with scissors. Few years after, Petitot et al. (2008, 2013) showed that expression of both CaWRKY1a (CaCc) and CaWRKY1b (CaCe) homeologs was upregulated in wounded leaves of C. arabica (see Sect. 7). In parallel, wounding also markedly upregulated expression of CaWRKY1a and CaWRKY1b genes in leaves of C. canephora and C. eugenioides, respectively, confirming that both genes were functional. In addition to CaWRKY1, Ramiro et al. (2010) also showed that CaWRKY19/20/21 genes, as well as CaWRKY15 and CaWRKY17, were also highly induced by wounding. In another work, Brandalise et al. (2009) showed that expression of CaIRL, coding an isoflavone reductase-like protein, was induced in leaves of C. arabica submitted to a mechanical injury, leading to further study the promoter of this gene (see Sect. 8).
6 Gene Expression in F1 Hybrids of C. arabica
In the context, the creation of new coffee varieties better adapted to biotic and abiotic stresses to low levels of inputs and to CC is now one of the challenges of several coffee research institutes (van der Vossen et al. 2015; Bertrand et al. 2019).
In C. arabica, it is possible to create and select in a relatively short time (e.g., around 8 years against 25 years for conventional breeding programs) new F1 hybrid varieties with increased production (e.g., under agroforestry) and also improved aromatic quality without increasing fertilizer quantities (Bertrand et al. 2006, 2011), by crossing pure commercial line varieties with phylogenetically distant plants corresponding to wild individuals from Ethiopia and Sudan, for example (Van der Vossen et al. 2015). The objective of the H2020 BREEDCAFSFootnote 2 (BREEDing Coffee for Agroforestry Systems) project, supported by the EU (2017–2021), is to identify robust markers (allelic, molecular, epigenetic) that could be used as early predictors to speed up future C. arabica breeding programs aiming to create new F1 hybrids with increased resistance and greater resilience to climate change in agroforestry systems (Bertrand et al. 2019). This project intends to compare the leaf transcriptomic profiles in F1 hybrids and cultivated varieties (and/or hybrids to their two parents) upon different abiotic stresses either performed in phytotrons and greenhouses (e.g., in order to test the effects of temperature, light, drought, CO2, and N2) or in field trials (or in networks of “demoplots” in farms). The numerous RNAseq studies planned to be perform within the framework of this project (Table 5) should also help us to better understand why the pure line varieties are less adapted to environmental constraints than F1 hybrids. For example, Toniutti et al. (2019b) showed that hybrid vigor (heterosis) could be explained by the modification of leaf expression profiles of several genes involved in the circadian clock (e.g., LHY and GIGANTEA), the chlorophyll synthesis (e.g., POR1A and POR1B), and starch degradation (e.g., CcGWD1 and CcISA3) in leaves of the C. arabica F1 hybrid GPFA124 compared to those of the inbred Caturra line. In the same work, upregulated expression of chloroplast genes in the C. arabica GPFA124 was also reported (see Sect. 7).
7 Expression of Chloroplast Genes
The chloroplast genome of C. arabica consists of 155,189 base pairs encoding 130 genes with 18 intron-containing genes (Samson et al. 2007). In a pioneer work, Dinh et al. (2016) analyzed the effects of drought, cold, or combined drought and heat stresses on intron splicing and expression patterns of 48 chloroplast genes from C. arabica. By RT-qPCR, these authors showed that the transcript levels of chloroplast mRNAs were globally decreased in seedlings submitted to drought or cold treatments. For example, expression of rbcL (coding the large subunit of Rubisco) and psaA and psaB (coding photosystem I proteins) was significantly reduced in C. arabica under cold stress conditions but not under drought. Regarding intron-containing genes, it was also shown that the splicing efficiencies of trnG, trnK, and trnA genes increased upon drought, combined drought and heat, or cold stress treatments, while these efficiencies decreased for trnL under these stresses. On the other hand, the splicing efficiencies of mRNA genes rps16, atpF, petB, and rpl2 were decreased upon drought but increased upon cold stress treatment.
Overexpression of CaPsbB gene (coding the photosystem II CP47 chlorophyll apoproteins) was also reported, either by in silico (Vieira et al. 2006; Mondego et al. 2011; Vinecky et al. 2012) or by in vivo (Mofatto et al. 2016) analyses, in leaves of drought-stressed coffee plants but also in those infected by H. vastatrix (Fernandez et al. 2012).
In addition to the circadian genes (see Sect. 6), Toniutti et al. (2019b) also reported increased photosynthetic electron transport efficiency in the C. arabica hybrid GPFA124 probably explained by higher expression of chloroplast genes CaPsbA and CaPsbD (coding the D1 and D2 proteins of PSII, respectively); CaPetA, CaPetD, and CaPetB (coding proteins of the cytB6/f complex); and CaPsaA, CaPsaB, and CaPsaJ (coding proteins of PSI), in this hybrid compared to the C. arabica cv. Caturra.
8 Coffee Promoters
The expression studies previously detailed also led to the identification of coffee promoters (De Almeida et al. 2008). For several of them, they were functionally characterized using the uidA (coding the β-glucuronidase) as the reporter gene by transgenic approaches either in Nicotiana tabacum or in Coffea sp. The first promoter was cloned from the CaCSP1 gene of C. arabica coding for the 11S seed storage protein and was shown to function as a bean (endosperm)-specific promoter in transgenic tobacco plants (Marraccini et al. 1999). A similar result was also observed for the shorter and medium promoter fragments of the CaLTP gene coding non-specific lipid transfer proteins (Cotta et al. 2014). Leaf-specific expression was also reported for CaRBCS1 and CcMXMT1 coffee promoters in transgenic tobacco (Marraccini et al. 2003; Satyanarayana et al. 2005). The SERK1 (somatic embryogenesis receptor-like kinase 1) promoter from C. canephora was also shown to drive the uidA expression in different embryo structures such as globular, heart, torpedo, and cotyledonal embryos present at 60 days after embryogenic induction (Jiménez-Guillen et al. 2018). Regarding abiotic stress, Brandalise et al. (2009) showed that the promoter of CaIRL was induced by wounded leaves of N. tabacum. In 2016, Nobres et al. analyzed the promoter function of the CaHB12 from C. arabica, a gene coding member of the homeodomain-leucine zipper I subfamily (HD-Zip) and conferring greater tolerance to drought stress when overexpressed in Arabidopsis (Alves-Ferreira et al. 2012). The study of transgenic A. thaliana plants bearing pCaHB12::GUS constructs showed that this promoter was expressed in leaves during drought and in roots after polyethylene glycol or mannitol treatments. On the other hand, the different haplotypes of the CcDREB1D promoter from C. canephora were shown to be upregulated by different abiotic stresses in the leaves of C. arabica (see Sect. 5.1) and N. tabacum transgenic plants (Alves et al. 2017, 2018; de Aquino et al. 2018). Regarding biotic stress, Petitot et al. (2013) analyzed the promoter activities of CaWRKY1a (named pW1a) and CaWRKY1b (named pW1b) homeologous genes, previously identified to be induced by CLR infestation in the C. arabica leaves (see Sects. 2.3, 4.1, and 5.6), in transient assays of N. benthamiana leaves, and in stable transgenic plants of C. arabica. These authors also showed increased activities of both promoters in leaves of tobacco treated with SA or in those of coffee infected with CLR, as well as increased activities of pW1a upon wounding. The other coffee promoters already described in the literature but without being tested in transgenic plants are cited in Table 6.
9 Coffee Small RNA (sRNA)
Using small (20 ± 26 nt) homologous sequences, small RNAs (sRNA) are known to play important roles by silencing pathways at the transcriptional or translational levels. Plant sRNAs are classified as (1) microRNAs (miRNAs) which are derived from self-complementary hairpin structures and (2) small interfering RNAs (siRNAs) which are derived from double-stranded RNA (dsRNA) or hairpin precursors (Borges and Martienssen 2015). The core mechanism of sRNA production requires the endonuclease activity of DICER-LIKE 1 (DCL1) and ARGONAUTE (AGO) proteins as effectors of silencing, while siRNA biogenesis involves action of RNA-dependent RNA polymerase (RDR), Pol IV, and Pol V. With the release of the C. canephora genome (Denoeud et al. 2014), sRNAs were now identified.
One of the first attempts to study coffee miRNAs was performed by Nellikunnumal and Chandrashekar (2012) who identified 18 miRNAs, belonging to 12 families, from C. canephora ESTs by computational approaches. By RT-PCR, these authors showed that expression was detected for seven families (viz., mir156, mir169, mir172, mir319, mir393, mir395, and mir396) in C. canephora leaves. By the same computational approach, Rebijith et al. (2013), Loss-Morais et al. (2014), and Devi et al. (2016) also identified miRNAs in C. arabica and C. canephora, showing that the majority of their potential targets corresponded to mRNA coding proteins involved in transcriptional regulation and signal transduction pathways. In another study, Akter et al. (2014) identified a potential miRNA (named mir393) from C. arabica ESTs and also showed that this sequence had as potential targets several genes coding transcription factors (e.g., bHLH7 and WRKY TFs) or proteins involved in auxin signaling pathway and plant defense responses (e.g., auxin signaling F-box 2 and auxin transporter protein 1). Using a specific pipeline to search for miRNA homologs on expressed sequence tag (EST) and genome survey sequence (GSS) coffee databases, Chaves et al. (2015) identified 36 microRNAs and a total of 616 and 362 potential target genes for C. arabica and C. canephora, respectively. Using a stem-loop RT-PCR assay, these authors also detected a higher amount of miRNAs (miRNAs 171, 172, 390, and 167) in leaves of C. arabica than in those of C. canephora, suggesting a possible role of sRNA in regulating C. arabica transcriptome.
Fernandes-Brum et al. (2017b) identified 11 AGO proteins, nine DCL-like proteins, eight RDR proteins, and 48 other proteins implicated in the sRNA pathways. These authors also identified (1) 235 miRNA precursors producing 317 mature miRNAs belonging to 113 MIR families and (2) 2239 putative C. canephora miRNA targets in different pathways. In another study, Bibi et al. (2017) also identified potential miRNAs potentially targeting 150 genes coding transcription factors but also proteins involved in multiple biological and metabolic processes, hypothetical proteins, signal transduction, transporters, growth and development, stress-related processes, structural constituents, and disease-related processes, for example.
In the study analyzing coffee memory to multiple drought exposures, de Freitas Guedes et al. (2018) also reported upregulated expression of mir398 and mir408 by the drought cycles in C. canephora. In addition to drought, these genes were also reported to be regulated in other plants by ABA, heat, UV, and also biotic stress events (Zhu et al. 2011; Khraiwesh et al. 2012; Guan et al. 2013). Interestingly, transgenic chickpea plants overexpressing mir408 were shown to be tolerant to several stresses including drought (Hajyzadeh et al. 2015; Ma et al. 2015). In the recent study, dos Santos et al. (2019) analyzed the transcriptome in N-starved roots of C. arabica and also identified 86 microRNA families targeting 253 genes. RT-qPCR assays showed that expression profiles of mir169, mir171, mir167, mir393, and mir858 were upregulated in roots after N-starvation, while mircar1 was downregulated after prolonged N-restriction. Altogether, these results highlight the role that might play sRNA in modulating the expression of genes involved in the adaptive responses of coffee plants to environmental factors.
10 Conclusions
Like many other crops, gene identification and characterization are of fundamental biological interest in coffee to understand the transcription networks involved in important agronomic traits and further to identify SNPs that can serve as markers of specific phenotypes to better drive future breeding programs. In that way, the high number of large-scale expression analyses, together with the recent access to long-read sequencing of transcripts (Cheng et al. 2017), to reference transcriptomes (Yuyama et al. 2016; Cheng et al. 2018), and to reference genomes of C. canephora (Denoeud et al. 2014) and C. arabica (de Kochko et al. 2015, 2017; Gaitan et al. 2015; Morgante et al. 2015; Yepes et al. 2016), now opens the way to identify SNPs associated with bean biochemical compound content (Tran et al. 2018) and adaptation to environmental factors (de Aquino et al. 2019) and to initiate marker-assisted selection (Alkimim et al. 2017) and genome-wide association studies (Andrade 2018; Sant’Ana et al. (2018); Carneiro et al. 2019). Together with the help of CRISPR/Cas9 technology (Breitler et al. 2018), it is now possible to greatly shorten the time required to create new coffee varieties with improved agronomic traits under CC.
Notes
- 1.
Gene names found in the Coffee Genome Hub (http://coffee-genome.org/)
- 2.
References
Abreu HMC, Nobile PM, Shimizu MM, Yamamoto PY, Silva EA, Colombo CA, Mazzafera P (2012) Influence of air temperature on proteinase activity and beverage quality in Coffea arabica. Braz J Bot 35:357–376
Acuña R, Bassüner R, Beillinson V, Cortina H, Cadena-Gómez G, Montes V, Nielsen N (1999) Coffee seeds contain 11S storage proteins. Physiol Plant 105:122–131
Aggarwal RK, Hendre PS, Varshney RK, Bhat PR, Krishnakumar V, Singh L (2007) Identification, characterization and utilization of EST-derived genic microsatellite markers for genome analyses of coffee and related species. Theor Appl Genet 114:359–372
Akter A, Islam MM, Mondal SI, Mahmud Z, Jewel NA, Ferdous S, Amin MR, Rahman MM (2014) Computational identification of miRNA and targets from expressed sequence tags of coffee (Coffea arabica). Saudi J Biol Sci 21:3–12
Alkimim ER, Caixeta ET, Sousa TV, Pereira AA, de Oliveira ACB, Zambolim L, Sakiyama NS (2017) Marker-assisted selection provides arabica coffee with genes from other Coffea species targeting on multiple resistance to rust and coffee berry disease. Mol Breeding 37:6
Alvarenga SM, Caixeta ET, Hufnagel B, Thiebaut F, Maciel-Zambolim E, Zambolim L, Sakiyama NS (2010) In silico identification of coffee genome expressed sequences potentially associated with resistance to diseases. Genet Mol Biol 33:795–806
Alves LC, de Magalhães DM, Labate MTV, Guidetti-Gonzalez S, Labate CA, Domingues DS, Sera T, Vieira LGE, Pereira LFP (2016) Differentially accumulated proteins in Coffea arabica seeds during perisperm tissue development and their relationship to coffee grain size. J Agric Food Chem 64:1635–1647
Alves GSC, Torres LF, Déchamp E, Breitler J-C, Joët T, Gatineau F, Andrade AC, Bertrand B, Marraccini P, Etienne H (2017) Differential fine-tuning of gene expression regulation in coffee leaves by CcDREB1D promoter haplotypes under water deficit. J Exp Bot 68:3017–3031
Alves GSC, Torres LF, de Aquino SO, Reichel T, Freire LP, Vieira NG, Vinecky F, This D, Pot D, Etienne H, Paiva LV, Marraccini P, Andrade AC (2018) Nucleotide diversity of the coding and promoter regions of DREB1D, a candidate gene for drought tolerance in Coffea species. Trop Plant Biol 11:31–48
Alves-Ferreira M,Waltenberg FPC, Pinto ERC, Grossi-de-Sá FM (2012) Use of the coffee homeobox gene CaHB12 to produce transgenic plants with greater tolerance to water scarcity and salt stress. WO061911:A2
Andrade AC (2018) Developments in molecular breeding techniques in Robusta coffee. In: Lashermes P (ed) Achieving sustainable cultivation of coffee: breeding and quality traits. Burleigh Dodds Science Publishing, Cambridge
Ashihara H (2006) Metabolism of alkaloids in coffee plants. Braz J Plant Physiol 18:1–8
Ashihara H, Monteiro AM, Gillies FM, Crozier A (1996) Biosynthesis of caffeine in leaves of coffee. Plant Physiol 111:747–753
Asquini E, Gerdol M, Gasperini D, Igic B, Graziosi G, Pallavicini A (2011) S-RNase-like sequences in styles of Coffea (Rubiaceae). Evidence for S-RNase based gametophytic self-incompatibility? Trop Plant Biol 4:237–249
Assad ED, Pinto HS, Zullo J Jr, AMH A (2004) Impacto das mudanças climáticas no zoneamento agroclimático do café no Brasil. Pesqui Agropecu Bras 39:1057–1064
Avelino J, Willocquet L, Savary S (2004) Effects of crop management patterns on coffee rust epidemics. Plant Pathol 53:541–547
Avelino J, Cristancho M, Georgiou S, Imbach P, Aguilar L, Bornemann G, Läderach P, Anzueto F, Hruska A, Morales C (2015) The coffee rust crises in Colombia and Central America (2008–2013): impacts, plausible causes and proposed solutions. Food Secur 7:303–321
Avila RT, Martins SCV, Sanglard LMVP, dos Santos MS, Menezes-Silva PE, Detman KC, Sanglard ML, Cardoso AA, Morais LE, Vital CE, Araújo WL, Nunes-Nesi A, DaMatta FM (2020) Starch accumulation does not lead to feedback photosynthetic downregulation in girdled coffee branches under varying source-to-sink ratios. Trees 34:1–16
Baba VY, Braghini MT, dos Santos TB, de Carvalho K, Soares JDM, Ivamoto-Suzuki ST, Maluf MP, Padilha L, Paccola-Meirelles LD, Pereira LF, Domingues DS (2020) Transcriptional patterns of Coffea arabica L. nitrate reductase, glutamine and asparagine synthetase genes are modulated under nitrogen suppression and coffee leaf rust. Peer J 8:e8320
Bang CF, Huyen TTT (2015) In silico identification, classification and expression analysis of genes encoding putative light-harvesting chlorophyll A/B-binding proteins in coffee (Coffea canephora L.). J Agric Technol 11:2547–2561
Barbosa BCF, Silva SC, de Oliveira RR, Chalfun-Junior A (2017) Zinc supply impacts on the relative expression of a metallothionein-like gene in Coffea arabica plants. Plant Soil 411:179–191
Bardil A, De Almeida JD, Combes MC, Lashermes P, Bertrand B (2011) Genomic expression dominance in the natural allopolyploid Coffea arabica is massively affected by growth temperature. New Phytol 192:760–774
Barreto HG, Lazzari F, Ságio SA, Chalfun-Junior A, Paiva LV (2012) In silico and quantitative analyses of the putative FLC-like homologue in coffee (Coffea arabica L.). Plant Mol Biol Rep 30:29–35
Barreto HG, Daude MM, Lima AA, Chalfun-Junior A (2018) Expression analysis of the coffee (Coffea arabica L.) FRIGIDA4-like gene (CaFRL4). Rev Desafios 5:204–213
Barsalobres-Cavallari CF, Severino FE, Maluf MP, Maia IG (2009) Identification of suitable internal control genes for expression studies in Coffea arabica under different experimental conditions. BMC Mol Biol 10:1
Batista-Santos P, Lidon FC, Fortunato A, Leitão AE, Lopes E, Partelli F, Ribeiro AI, Ramalho JC (2011) The impact of cold on photosynthesis in genotypes of Coffea spp. – photosystem sensitivity, photoprotective mechanisms and gene expression. J Plant Physiol 168:792–806
Bazzo BR, Eiras AL, DeLaat DM, Siqueira WJ, Mondego JMC, Colombo CA (2013) Gene expression analysis suggests temporal differential response to aluminum in Coffea arabica cultivars. Trop Plant Biol 6:191–198
Benchabane M, Schluter U, Vorster J, Goulet MC, Michaud D (2010) Plant cystatins. Biochimie 92:1657–1666
Berthaud J (1980) L’incompatibilité chez Coffea canephora: méthode de test et déterminisme génétique. Café Cacao Thé 24:267–274
Bertrand C, Noirot M, Doulbeau S, de Kochko A, Hamon S, Campa C (2003) Chlorogenic acid content swap during fruit maturation in Coffea pseudozanguebariae qualitative comparison with leaves. Plant Sci 165:1355–1361
Bertrand B, Vaast P, Alpizar E, Etienne H, Davrieux F, Charmetant P (2006) Comparison of bean biochemical composition and beverage quality of arabica hybrids involving Sudanese-Ethiopian origins with traditional varieties at various elevations in Central America. Tree Physiol 26:1239–1248
Bertrand B, Alpizar E, Lara L, Santacreo R, Hidalgo M, Quijano JM, Montagnon C, Georget F, Etienne H (2011) Performance of Coffea arabica F1 hybrids in agroforestry and full-sun cropping systems in comparison with American pure line cultivars. Euphytica 181:147–158
Bertrand B, Bardil A, Baraille H, Dussert S, Doulbeau S, Dubois E, Severac D, Dereeper A, Etienne H (2015) The greater phenotypic homeostasis of the allopolyploid Coffea arabica improved the transcriptional homeostasis over that of both diploid parents. Plant Cell Physiol 56:2035–2051
Bertrand B, Breitler J-C, Georget F, Penot E, Bordeaux M, Marraccini P, Léran S, Campa C, Bonato O, Villain L, Etienne H (2019) New varieties for innovative agroforestry coffee systems. In: Côte F-X, Poirier-Magona E, Perret S, Roudier P, Rapidel B, Thirion M-C (eds) The agroecological transition of agricultural systems in the global south. Versailles, Quae, pp 161–176
Bharathi K, Sreenath HL (2017) Identification and analysis of jasmonate pathway genes in Coffea canephora (Robusta coffee) by in silico approach. Pharmacogn Mag 13:196–200
Bharathi K, Santosh P, Sreenath HL (2017) Transcripts of pectin-degrading enzymes and isolation of complete cDNA sequence of a pectate lyase gene induced by coffee white stem borer (Xylotrechus quadripes) in the bark tissue of Coffea canephora (robusta coffee). 3 Biotech 7:45
Bibi F, Barozai MYK, Din M (2017) Bioinformatics profiling and characterization of potential microRNAs and their targets in the genus Coffea. Turk J Agric For 41:191–200
Borges F, Martienssen RA (2015) The expanding world of small RNAs in plants. Nat Rev Mol Cell Biol 16:727–741
Bottcher A, Nobile PM, Martins PF, Conte FF, Azevedo RA, Mazzafera P (2011) A role for ferritin in the antioxidant system in coffee cell cultures. Biometals 24:225–237
Brandalise M, Severino FE, Maluf MP, Maia IG (2009) The promoter of a gene encoding an isoflavone reductase-like protein in coffee (Coffea arabica) drives a stress-responsive expression in leaves. Plant Cell Rep 28:1699–1708
Breitler J-C, Déchamp E, Campa C, Rodrigues LAZ, Guyot R, Marraccini P, Etienne H (2018) CRISPR/Cas9-mediated efficient targeted mutagenesis has the potential to accelerate the domestication of Coffea canephora. Plant Cell Tissue Organ Cult 134:383–394
Breitler J-C, Djerrab D, Leran S, Toniutti L, Guittin C, Severac D, Pratlong M, Dereeper A, Etienne H, Bertrand B (2020) Full moonlight-induced circadian clock entrainment in Coffea arabica. BMC Plant Biol 20:24
Budzinski IGF, Santos TB, Sera T, Pot D, Vieira LGE, Pereira LFP (2010) Expression patterns of three α-expansin isoforms in Coffea arabica during fruit development. Plant Biol 13:462–471
Bunn C, Läderach P, Ovalle Rivera O, Kirschke D (2015a) A bitter cup: climate change profile of global production of Arabica and Robusta coffee. Clim Chang 129:89–101
Bunn C, Läderach P, Pérez Jimenez JG, Montagnon C, Schilling T (2015b) Multiclass classification of agro-ecological zones for Arabica coffee: an improved understanding of the impacts of climate change. PLoS One 10:e0140490
Bustin SA (2002) Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): trends and problems. J Mol Endocrinol 29:23–39
Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT (2009) The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55:611–622
Bytof G, Knopp SE, Kramer D, Breitenstein B, Bergervoet JHW, Groot SPC, Selmar D (2007) Transient occurrence of seed germination processes during coffee post-harvest treatment. Ann Bot 100:61–66
Cação SMB, Leite TF, Budzinski IGF, dos Santos TB, Scholz MBS, Carpentieri-Pipolo V, Domingues DS, Vieira LGE, Pereira LFP (2012) Gene expression and enzymatic activity of pectin methylesterase during fruit development and ripening in Coffea arabica L. Genet Mol Res 11:3186–3197
Cacas J-L, Petitot A-S, Bernier L, Estevan J, Conejero G, Mongrand S, Fernandez D (2011) Identification and characterization of the non-race specific disease resistance 1 (NDR1) orthologous protein in coffee. BMC Plant Biol 11:144
Campa C, Ballester JF, Doulbeau S, Dussert S, Hamon S, Noirot M (2004) Trigonelline and sucrose diversity in wild Coffea species. Food Chem 88:39–44
Campa C, Urban L, Mondolot L, Fabre D, Roques S, Lizzi Y, Aarrouf J, Doulbeau S, Breitler J-C, Letrez C, Toniutti L, Bertrand B, La Fisca P, Bidel LPR, Etienne H (2017) Juvenile coffee leaves acclimated to low light are unable to cope with a moderate light increase. Front Plant Sci 8:1126
Cardoso DC, Martinati JC, Giachetto PF, Vidal RO, Carazzolle MF, Padilha L, Guerreiro-Filho O, Maluf MP (2014) Large-scale analysis of differential gene expression in coffee genotypes resistant and susceptible to leaf miner-toward the identification of candidate genes for marker assisted-selection. BMC Genomics 15:66
Carneiro FA, de Aquino SO, Mattos NG, Valeriano JC, Carneiro WWJ, Rodrigues GC, Carvalho MAF, Veiga AD, Grattapaglia D, Silva-Júnior OB, Marraccini P, Junior ICV, Balestre M, Andrade AC (2019) Genome wide selection in Coffea canephora breeding. In: X Simpósio de Pesquisa dos Cafés do Brasil Vitória (ES)-Brazil. EMBRAPA, Vitória
Cenci A, Combes MC, Lashermes P (2012) Genome evolution in diploid and tetraploid Coffea species. Plant Mol Biol 78:135–145
Chaves SS, Fernandes-Brum CN, Silva GFF, Ferrara-Barbosa BC, Paiva LV, Nogueira FTS, Cardoso TCS, Amaral LR, de Souza Gomes M, Chalfun-Junior A (2015) New insights on Coffea miRNAs: features and evolutionary conservation. Appl Biochem Biotechnol 177:879–908
Cheng B, Furtado A, Henry RJ (2017) Long-read sequencing of the coffee bean transcriptome reveals the diversity of full-length transcripts. Gigascience 6:1–13
Cheng B, Furtado A, Henry RJ (2018) The coffee bean transcriptome explains the accumulation of the major bean components through ripening. Sci Rep 8:11414
Cheserek JJ, Gichimu BM (2012) Drought and heat tolerance in coffee: a review. Int Res J Agric Sci Soil Sci 2:498–501
Combes MC, Cenci A, Baraille H, Bertrand B, Lashermes P (2012) Homeologous gene expression in response to growing temperature in a recent allopolyploid (Coffea arabica L.). J Hered 103:36–46
Combes M-C, Dereeper A, Severac D, Bertrand B, Lashermes P (2013) Contribution of subgenomes to the transcriptome and their intertwined regulation in the allopolyploid Coffea arabica grown at contrasted temperatures. New Phytol 200:251–260
Combes M-C, Hueber Y, Dereeper A, Rialle S, Herrera J-C, Lashermes P (2015) Regulatory divergence between parental alleles determines gene expression patterns in hybrids. Genome Biol Evol 7:1110–1121
Costa TS (2014) Análise do perfil transcriptômico e proteômico de raízes de diferentes clones de Coffea canephora em condições de déficit hídrico. PhD dissertation, Federal University of Lavras, Brazil
Costa TS, Melo JAT, Carneiro FA, Vieira NG, Rêgo ECS, Block C Jr, Marraccini P, Andrade AC (2015) Drought effects on expression of genes involved in ABA signaling pathway in roots of susceptible and tolerant clones of C. canephora. In: 25th international conference on coffee science (ASIC), Armenia, Colombia (PB236), pp 42–45
Cotta MG (2017) Molecular mechanisms in the first step of ABA-mediated response in Coffea ssp. PhD dissertation, SupAgro, Montpellier, France, p 176
Cotta MG, Barros LMG, De Almeida JD, De Lamotte F, Barbosa EA, Vieira NG, Alves GSC, Vinecky F, Andrade AC, Marraccini P (2014) Lipid transfer proteins in coffee: isolation of Coffea orthologs, Coffea arabica homeologs, expression during coffee fruit development and promoter analysis in transgenic tobacco plants. Plant Mol Biol 85:11–31
Couttolenc-Brenis E, Carrión GL, Villain L, Ortega-Escalona F, Ramírez-Martínez D, Mata-Rosas M, Méndez-Bravo A (2020) Prehaustorial local resistance to coffee leaf rust in a Mexican cultivar involves expression of salicylic acid-responsive genes. PeerJ 8:e8345
Cruz F, Kalaoun S, Nobile P, Colombo C, Almeida J, Barros LMG, Romano E, Grossi-De-Sá MF, Vaslin M, Alves-Ferreira M (2009) Evaluation of coffee reference genes for relative expression studies by quantitative real-time RT-PCR. Mol Breed 23:607–616
da Silva EAA, Acencio ML, Bovolenta LA, Lemke N, Varani AM, Bravo JP, Hoshino-Bezerra AA, Lemos EGM (2019) Gene expression during the germination of coffee seed. J Seed Sci 41:168–179
DaMatta FM, Ramalho JDC (2006) Impacts of drought and temperature stress on coffee physiology and production: a review. Braz J Plant Physiol 18:55–81
Davis AP, Gole TW, Baena S, Moat J (2012) The impact of climate change on indigenous arabica coffee (Coffea arabica): predicting future trends and identifying priorities. PLoS One 7:e47981
Davis AP, Chadburn H, Moat J, O’Sullivan R, Hargreaves S, Nic Lughadha E (2019) High extinction risk for wild coffee species and implications for coffee sector sustainability. Sci Adv 5:eaav3473
de Almeida JD, Barros LMG, Santos DBM, Cotta MG, Barbosa EA, Cação SB, Eira MTS, Alves GSC, Vinecky F, Pereira LFP, da Silva FR, Andrade AC, Marraccini P, Carneiro M (2008) Prospection of tissue specific promoters in coffee. In: 22th international conference on coffee science (ASIC), Campinas, Brazil, pp 954–957
de Aquino SO, Carneiro FA, Rêgo ECS, Alves GSC, Andrade AC, Marraccini P (2018) Functional analysis of different promoter haplotypes of the coffee (Coffea canephora) CcDREB1D gene through genetic transformation of Nicotiana tabacum. Plant Cell Tissue Organ Cult 132:279–294
de Aquino SO, Tournebize R, Marraccini P, Mariac C, Bethune K, Andrade AC, Kiwuka C, Crouzillat D, Anten NPR, de Kochko A, Poncet V (2019) Towards the identification of candidate gene nucleic polymorphisms to predict the adaptedness of Ugandense C. canephora populations to climate change. In: 27th international conference on coffee science (ASIC), Portland (OR), USA
de Carvalho K, Bespalhok Filho JC, dos Santos TB, de Souza SGH, Vieira LGE, Pereira LFP, Domingues DS (2013) Nitrogen starvation, salt and heat stress in coffee (Coffea arabica L.): identification and validation of new genes for qPCR normalization. Mol Biotechnol 53:315–325
de Carvalho K, Petkowicz CLO, Nagashima GT, Bespalhok Filho JC, Vieira LGE, Pereira LFP, Domingues DS (2014) Homeologous genes involved in mannitol synthesis reveal unequal contributions in response to abiotic stress in Coffea arabica. Mol Gen Genomics 289:951–963
De Castro R, Marraccini P (2006) Cytology, biochemistry and molecular changes during coffee fruit development. Braz J Plant Physiol 18:175–199
de Freitas Guedes FA, Nobres P, Ferreira DCR, Menezes-Silva PE, Ribeiro-Alves M, Correa RL, DaMatta FM, Alves-Ferreira M (2018) Transcriptional memory contributes to drought tolerance in coffee (Coffea canephora) plants. Environ Exp Bot 147:220–233
de Kochko A, Akaffou S, Andrade AC, Campa C, Crouzillat D, Guyot R, Hamon P, Ming R, Mueller LA, Poncet V, Tranchant Dubreuil C, Hamon S (2010) Advances in Coffea genomics. In: Kader JC, Delseny M (eds) Advances in botanical research, vol 53, pp 23–63
de Kochko A, Crouzillat D, Rigoreau M, Lepelley M, Bellanger L, Merot-L’Anthoene V, Vandecasteele C, Guyot R, Poncet V, Tranchant-Dubreuil C, Hamon P, Hamon S, Couturon E, Descombes P, Moine D, Mueller L, Strickler SR, Andrade AC, Protasio LFP, Marraccini P, Giuliano G, Fiore A, Pietrella M, Aprea G, Ming R, Wai J, Domingues DS, Paschoal A, Kuhn G, Korlach J, Chin J, Sankoff D, Zheng C, Albert VA (2015) Dihaploid Coffea arabica genome sequencing and assembly. In: Plant and animal genome XXIII. San Diego (CA), USA. https://pag.confex.com/pag/xxiii/webprogram/Paper16983.html
de Kochko A, Hamon S, Guyot R, Couturon E, Poncet V, Dubreuil-Tranchant C, Crouzillat D, Rigoreau M, Hamon P (2017) Omics applications: coffee. In: Chowdappa P, Karun A, Rajesh MK, Ramesh SV (eds) Biotechnology of plantation crops. Daya Publishing House, New Delhi, pp 589–606
De Nardi B, Dreos R, Del Terra L, Martellossi C, Asquini E, Tornincasa P, Gasperini D, Pacchioni B, Rathinavelu R, Pallavicini A, Graziosi G (2006) Differential responses of Coffea arabica L. leaves and roots to chemically induced systemic acquired resistance. Genome 49:1594–1605
de Oliveira RR, Chalfun-Junior A, Paiva LV, Andrade AC (2010) In silico and quantitative analyses of MADS-box genes in Coffea arabica. Plant Mol Biol Rep 28:460–472
de Oliveira RR, Cesarino I, Mazzafera P, Dornelas MC (2014) Flower development in Coffea arabica L.: new insights into MADS-box genes. Plant Reprod 27:79–94
de Oliveira KC, de Souza GP, Bazioli JM, Martinati JC, dos Santos MM, Padilha L, Guerreiro-Filho O, Maluf MP (2019) Effects of somatic embryogenesis on gene expression of cloned coffee heterozygous hybrids. Acta Physiol Plant 41:118
Denoeud F, Carretero-Paulet L, Dereeper A, Droc G, Guyot R, Pietrella M, Zheng C, Alberti A, Anthony F, Aprea G, Aury J-M, Bento P, Bernard M, Bocs S, Campa C, Cenci A, Combes M-C, Crouzillat D, Da Silva C, Daddiego L, De Bellis F, Dussert S, Garsmeur O, Gayraud T, Guignon V, Jahn K, Jamilloux V, Joet T, Labadie K, Lan T, Leclercq J, Lepelley M, Leroy T, Li L-T, Librado P, Lopez L, Munoz A, Noel B, Pallavicini A, Perrotta G, Poncet V, Pot D, Priyono RM, Rouard M, Rozas J, Tranchant-Dubreuil C, VanBuren R, Zhang Q, Andrade AC, Argout X, Bertrand B, de Kochko A, Graziosi G, Henry RJ, Jayarama MR, Nagai C, Rounsley S, Sankoff D, Giuliano G, Albert VA, Wincker P, Lashermes P (2014) The coffee genome provides insight into the convergent evolution of caffeine biosynthesis. Science 345:1181–1184
Devi KJ, Chakraborty S, Deb B, Rajwanshi R (2016) Computational identification and functional annotation of microRNAs and their targets from expressed sequence tags (ESTs) and genome survey sequences (GSSs) of coffee (Coffea arabica L.). Plant Gene 6:30–42
Dinh SN, Sai TZT, Nawaz G, Lee K, Kang H (2016) Abiotic stresses affect differently the intron splicing and expression of chloroplast genes in coffee plants (Coffea arabica) and rice (Oryza sativa). J Plant Physiol 201:85–94
Diniz I, Figueiredo A, Loureiro A, Batista D, Azinheira H, Várzea V, Pereira AP, Gichuru E, Moncada P, Guerra-Guimarães L, Oliveira H, Silva MDC (2017) A first insight into the involvement of phytohormones pathways in coffee resistance and susceptibility to Colletotrichum kahawae. PLoS One 12:e0178159
Diola V, Brito GG, Caixeta ET, Pereira LFP, Loureiro ME (2013) A new set of differentially expressed signaling genes is early expressed in coffee leaf rust race II incompatible interaction. Funct Integr Genomics 13:379–389
Dong X, Jiang Y, Yang Y, Xiao Z, Bai X, Gao J, Tan S, Hur Y, Hao S, He F (2019a) Identification and expression analysis of the NAC gene family in Coffea canephora. Agronomy 9:670
Dong X, Yang Y, Zhang Z, Xiao Z, Bai X, Gao J, Hur Y, Hao S, He F (2019b) Genome-wide identification of WRKY genes and their response to cold stress in Coffea canephora. Forests 10:335
dos Santos AB, Mazzafera P (2013) Aquaporins and the control of the water status in coffee plants. Theor Exp Plant Physiol 25:79–93
dos Santos TB, Budzinski IGF, Marur CJ, Petkowicz CLO, Pereira LFP, Vieira LGE (2011) Expression of three galactinol synthase isoforms in Coffea arabica L. and accumulation of raffinose and stachyose in response to abiotic stresses. Plant Physiol Biochem 49:441–448
dos Santos TB, de Lima RB, Nagashima GT, Petkowicz CLO, Carpentieri-Pipolo V, Pereira LFP, Domingues DS, Vieira LGE (2015) Galactinol synthase transcriptional profile in two genotypes of Coffea canephora with contrasting tolerance to drought. Genet Mol Biol 38:182–190
dos Santos TB, Lima JE, Felicio MS, Soares JDM, Domingues DS (2017) Genome-wide identification, classification and transcriptional analysis of nitrate and ammonium transporters in Coffea. Genet Mol Biol 40:346–359
dos Santos TB, Soares JDM, Lima JE, Silva JC, Ivamoto ST, Baba VY, Souza SGH, Lorenzetti APR, Paschoal AR, Meda AR, Nishiyama Júnior MY, de Oliveira ÚC, Mokochinski JB, Guyot R, Junqueira-de-Azevedo ILM, Figueira AVO, Mazzafera P, Júnior OR, Vieira LGE, Pereira LFP, Domingues DS (2019) An integrated analysis of mRNA and sRNA transcriptional profiles in Coffea arabica L. roots: insights on nitrogen starvation responses. Funct Integr Genomics 19:151–169
Dussert S, Serret J, Bastos-Siqueira A, Morcillo F, Déchamp E, Rofidal V, Lashermes P, Etienne H, Joët T (2018) Integrative analysis of the late maturation programme and desiccation tolerance mechanisms in intermediate coffee seeds. J Exp Bot 69:1583–1597
Echeverria-Beirute F, Murray SC, Klein P, Kerth C, Miller R, Bertrand B (2018) Rust and thinning management effect on cup quality and plant performance for two cultivars of Coffea arabica L. J Agric Food Chem 66:5281–5292
Echeverría-Beirute F, Murray SC, Bertrand B, Klein PE (2019) Candidate genes in coffee (Coffea arabica L.) leaves associated with rust (Hemileia vastatrix Berk. & Br) stress. Peer J Preprints 7:e27923v1
Eira MTS, Amaral da Silva EA, de Castro RD, Dussert S, Walters C, Bewley JD, Hilhorst HWM (2006) Coffee seed physiology. Braz J Plant Physiol 18:149–163
Eskes A, Leroy T (2004) Coffee selection and breeding. In: Wintgens JN (ed) Coffee: growing, processing, sustainable production. A guidebook for growers, processors, traders, and researchers. Wiley, Weinheim, pp 57–86
Etienne H, Breton D, Breitler J-C, Bertrand B, Déchamp E, Awada R, Marraccini P, Léran S, Alpizar E, Campa C, Courtel P, Georget F, Ducos J-P (2018) Coffee somatic embryogenesis: how did research, experience gained and innovations promote the commercial propagation of elite clones from the two cultivated species? Front Plant Sci 9:1630
Eulgem T (2006) Dissecting the WRKY web of plant defense regulators. PLoS Pathog 2:1028–1030
Falco MC, Marbach PAS, Pompermayer P, Lopes FCC, Silva-Filho MC (2001) Mechanisms of sugarcane response to herbivory. Genet Mol Biol 24:113–122
Fernandes-Brum CN, Garcia BO, Moreira RO, Sagio SA, Barreto HG, Lima AA, Freitas NG, Lima RR, Carvalho CHS, Chalfun-Junior A (2017a) A panel of the most suitable reference genes for RT-qPCR expression studies of coffee: screening their stability under different conditions. Tree Genet Genomes 13:131
Fernandes-Brum CN, Rezende PM, Ribeiro THC, de Oliveira RR, Cardoso TCS, Amaral LR, Gomes MS, Chalfun-Junior A (2017b) A genome-wide analysis of the RNA-guided silencing pathway in coffee reveals insights into its regulatory mechanisms. PLoS One 12:e0176333
Fernandez D, Santos P, Agostini C, Bon MC, Petitot AS, Silva MC, Guerra-Guimarães L, Ribeiro A, Argout X, Nicole M (2004) Coffee (Coffea arabica L.) genes early expressed during infection by the rust fungus (Hemileia vastatrix). Mol Plant Pathol 5:527–536
Fernandez D, Tisserant E, Talhinhas P, Azinheira H, Vieira A, Petitot A-S, Loureiro A, Poulain J, Da Silva C, Silva MC, Duplessis S (2012) 454-pyrosequencing of Coffea arabica leaves infected by the rust fungus Hemileia vastatrix reveals in planta-expressed pathogen-secreted proteins and plant functions in a late compatible plant-rust interaction. Mol Plant Pathol 13:17–37
Figueiredo SA, Lashermes P, Aragão FJ (2011) Molecular characterization and functional analysis of the β-galactosidase gene during Coffea arabica (L.) fruit development. J Exp Bot 62:2691–2703
Figueiredo A, Loureiro A, Batista D, Monteiro F, Várzea V, Pais MS, Gichuru EK, Silva MC (2013) Validation of reference genes for normalization of qPCR gene expression data from Coffea spp. hypocotyls inoculated with Colletotrichum kahawae. BMC Res Notes 6:388
Florez JC, Mofatto LS, do Livramento Freitas-Lopes R, Ferreira SS, Zambolim EM, Carazzolle MF, Zambolim L, Caixeta ET (2017) High throughput transcriptome analysis of coffee reveals prehaustorial resistance in response to Hemileia vastatrix infection. Plant Mol Biol 95:607–623
Fortunato AS, Lidon FC, Batista-Santos P, Leitão AE, Pais IP, Ribeiro AI, Ramalho JC (2010) Biochemical and molecular characterization of the antioxidative system of Coffea sp. under cold conditions in genotypes with contrasting tolerance. J Plant Physiol 167:333–342
Freire LP, Marraccini P, Rodrigues GC, Andrade AC (2013) Analysis of the mannose 6 phosphate reductase gene expression in coffee trees submitted to water deficit. Coffee Sci 8:17–23
Freitas NC, Barreto HG, Fernandes-Brum CN, Moreira RO, Chalfun-Junior A, Paiva LV (2017) Validation of reference genes for qPCR analysis of Coffea arabica L. somatic embryogenesis-related tissues. Plant Cell Tissue Organ Cult 128:663–678
Frischknecht PM, Ulmer-Dufek J, Baumann TW (1986) Purine alkaloid formation in buds and developing leaflets of Coffea arabica: expression of an optimal defense strategy? Phytochemistry 25:613–616
Gaborit C, Caillet V, Deshayes A, Marraccini P (2003) Molecular cloning of a full-length cDNA and gene from Coffea arabica encoding a protein homologous to the yeast translation initiation factor SUI1: expression analysis in plant organs. Braz J Plant Physiol 15:55–58
Gaitan A, Cristancho MA, Gongora CE, Moncada P, Posada H, Gast F, Yepes M, Aldwinckle H (2015) Long-read deep sequencing and assembly of the allotetraploid Coffea arabica cv. Caturra and its maternal ancestral diploid species Coffea eugenioides. In: Plant and animal genome XXIII. San Diego, CA (USA). https://pag.confex.com/pag/xxiii/webprogram/Paper17662.html
Ganesh D, Petitot A-S, Silva MC, Alary R, Lecouls A-C, Fernandez D (2006) Monitoring of the early molecular resistance responses of coffee (Coffea arabica L.) to the rust fungus (Hemileia vastatrix) using real-time quantitative RT-PCR. Plant Sci 170:1045–1051
Gaspari-Pezzopane C, Bonturi N, Guerreiro Filho O, Favarin JL, Maluf MP (2012) Gene expression profile during coffee fruit development and identification of candidate markers for phenological stages. Pesqui Agropecu Bras 47:972–982
Georget F, Marie L, Alpizar E, Courtel P, Bordeaux M, Hidalgo JM, Marraccini P, Breitler J-C, Déchamp E, Poncon C, Etienne H, Bertrand B (2019) Starmaya: the first Arabica F1 coffee hybrid produced using genetic male sterility. Front Plant Sci 10:1344
Geromel C, Ferreira LP, Guerreiro SMC, Cavalari AA, Pot D, Pereira LFP, Leroy T, Vieira LGE, Mazzafera P, Marraccini P (2006) Biochemical and genomic analysis of sucrose metabolism during coffee (Coffea arabica) fruit development. J Exp Bot 57:3243–3258
Geromel C, Ferreira LP, Bottcher A, Pot D, Pereira LFP, Leroy T, Vieira LGE, Mazzafera P, Marraccini P (2008a) Sucrose metabolism during fruit development in Coffea racemosa. Ann Appl Biol 152:179–187
Geromel C, Ferreira LP, Davrieux F, Guyot B, Ribeyre F, dos Santos Scholz MB, Pereira LFP, Vaast P, Pot D, Leroy T, Androcioli Filho A, Vieira LGE, Mazzafera P, Marraccini P (2008b) Effects of shade on the development and sugar metabolism of coffee fruits. Plant Physiol Biochem 46:569–579
Ghini R, Hamada E, Pedro Júnior MJ, Marengo JA, Gonçalves RRV (2008) Risk analysis of climate change on coffee nematodes and leaf miner in Brazil. Pesqui Agropecu Bras 43:187–194
Ghini R, Hamada E, Pedro Júnior MJ, Gonçalves RRV (2011) Incubation period of Hemileia vastatrix in coffee plants in Brazil simulated under climate change. Summa Phytopathol 37:85–93
Ghini R, Torre-Neto A, Dentzien AFM, Guerreiro-Filho O, Iost R, FRA P, JSM P, Thomaziello RA, Bettiol W, FM DM (2015) Coffee growth, pest and yield responses to free-air CO2 enrichment. Clim Chang 132:307–320
Gichuru EK (1997) Resistance mechanisms in Arabica coffee to coffee berry disease Colletotrichum kahawae sp. nov-a review. Kenya Coffee 727:2441–2445
Gichuru EK (2007) Histological comparison of susceptible and resistant interactions of coffee (Coffea arabica and C. canephora varieties) and Colletotrichum kahawae. Agron Afr 19:233–240
González-Mendoza VM, Sánchez-Sandoval ME, Munnik T, Hernández-Sotomayor SMT (2020) Biochemical characterization of phospholipases C from Coffea arabica in response to aluminium stress. J Inorg Biochem 204:110951
Goulao LF, Fortunato AS, Ramalho JC (2012) Selection of reference genes for normalizing quantitative real-time PCR gene expression data with multiple variables in Coffea spp. Plant Mol Biol Rep 30:741–759
Grover CE, Gallagher JP, Szadkowski EP, Yoo M-J, Flagel LE, Wendel JF (2012) Homoeolog expression bias and expression level dominance in allopolyploids. New Phytol 196:966–971
Guan Q, Lu X, Zeng H, Zhang Y, Zhu J (2013) Heat stress induction of miR398 triggers a regulatory loop that is critical for thermotolerance in Arabidopsis. Plant J 74:840–851
Guerreiro-Filho O (2006) Coffee leaf miner resistance. Braz J Plant Physiol 18:109–117
Guerreiro-Filho O, Mazzafera P (2000) Caffeine does not protect coffee against the leaf miner Perileucoptera coffeella. J Chem Ecol 26:1447–1464
Guerreiro-Filho O, Silvarolla MB, Eskes AB (1999) Expression and mode of inheritance of resistance in coffee to leaf miner Perileucoptera coffeella. Euphytica 105:7–15
Haile M, Kang WH (2018) Transcriptome profiling of the coffee (C. arabica L.) seedlings under salt stress condition. J Plant Biotechnol 45:45–54
Hajyzadeh M, Turktas M, Khawar KM, Unver T (2015) miR408 overexpression causes increased drought tolerance in chickpea. Gene 555:186–193
Hinniger C, Caillet V, Michoux F, Ben Amor M, Tanksley S, Lin C, McCarthy J (2006) Isolation and characterization of cDNA encoding three dehydrins expressed during Coffea canephora (Robusta) grain development. Ann Bot 97:755–765
ICO (2020) Coffee production by exporting countries (data at July 2020). www.ico.org/prices/po-production.pdf
Idárraga SM, Castro AM, Macea EP, Gaitán AL, Rivera LF, Cristancho MA, Góngora CE (2012) Sequences and transcriptional analysis of Coffea arabica var. Caturra and Coffea liberica plant responses to coffee berry borer Hypothenemus hampei (Coleoptera: Curculionidae: Scolytinae) attack. J Plant Interact 7:56–70
IPCC (2013) Climate change 2013: the physical science basis. In: Stocker TF, Qin D, Plattner G-K, Tignor M, Allen SK, Boschung J, Nauels A, Xia Y, Bex V, Midgley PM (eds) Contribution of working group i to the fifth assessment report of the intergovernmental panel on climate change. Cambridge University Press, Cambridge, New York, p 1535
Ivamoto ST, Reis O Jr, Domingues DS, dos Santos TB, de Oliveira FF, Pot D, Leroy T, Vieira LGE, Carazzolle MF, Pereira GAG, Pereira LFP (2017a) Transcriptome analysis of leaves, flowers and fruits perisperm of Coffea arabica L. reveals the differential expression of genes involved in raffinose biosynthesis. PLoS One 12:e0169595
Ivamoto ST, Sakuray LM, Ferreira LP, Kitzberger CSG, Scholz MBS, Pot D, Leroy T, Vieira LGE, Domingues DS, Pereira LFP (2017b) Diterpenes biochemical profile and transcriptional analysis of cytochrome P450s genes in leaves, roots, flowers, and during Coffea arabica L. fruit development. Plant Physiol Biochem 111:340–347
Jaramillo J, Muchugu E, Vega FE, Davis A, Borgemeister C, Chabi-Olaye A (2011) Some like it hot: the influence and implications of climate change on coffee berry borer (Hypothenemus hampei) and coffee production in East Africa. PLoS One 6:e24528
Jiménez-Guillen D, Pérez-Pascual D, Souza-Perera R, Godoy-Hernández G, Zúñiga-Aguilar JJ (2018) Cloning of the Coffea canephora SERK1 promoter and its molecular analysis during the cell-to embryo transition. Electron J Biotechnol 36:34–46
Joët T, Laffargue A, Salmona J, Doulbeau S, Descroix F, Bertrand B, de Kochko A, Dussert S (2009) Metabolic pathways in tropical dicotyledonous albuminous seeds: Coffea arabica as a case study. New Phytol 182:146–162
Joët T, Salmona J, Laffargue A, Descroix F, Dussert S (2010) Use of the growing environment as a source of variation to identify the quantitative trait transcripts and modules of co-expressed genes that determine chlorogenic acid accumulation. Plant Cell Environ 33:1220–1233
Joët T, Pot D, Ferreira LP, Dussert S, Marraccini P (2012) Identification des déterminants moléculaires de la qualité du café par des approches de génomique fonctionnelle. Une revue. Cah Agric 21:125–133
Joët T, Laffargue A, Salmona J, Doulbeau S, Descroix F, Bertrand B, Lashermes P, Dussert S (2014) Regulation of galactomannan biosynthesis in coffee seeds. J Exp Bot 65:323–337
Kakegawa H, Shitan N, Kusano H, Ogita S, Yazaki K, Sugiyama A (2019) Uptake of adenine by purine permeases of Coffea canephora. Biosci Biotechnol Biochem 83:1300–1305
Kerepesi I, Galiba G (2000) Osmotic and salt stress induced alteration in soluble carbohydrate content in wheat seedlings. Crop Sci 40:482–487
Khraiwesh B, Zhu JK, Zhu J (2012) Role of miRNAs and siRNAs in biotic and abiotic stress responses of plants. Biochim Biophys Acta 1819:137–148
Kiba A, Saitoh H, Nishihara M, Omiya K, Yamamura S (2003) C-terminal domain of a hevein-like protein from Wasabia japonica has potent antimicrobial activity. Plant Cell Physiol 44:296–303
Klingler JP, Batelli G, Zhu JK (2010) ABA receptors: the START of a new paradigm in phytohormone signalling. J Exp Bot 61:3199–3210
Koshiro Y, Zheng XQ, Wang ML, Nagai C, Ashihara H (2006) Changes in content and biosynthetic activity of caffeine and trigonelline during growth and ripening of Coffea arabica and Coffea canephora fruits. Plant Sci 171:242–250
Kramer D, Breitenstein B, Kleinwächter M, Selmar D (2010) Stress metabolism in green coffee beans (Coffea arabica L.): expression of dehydrins and accumulation of GABA during drying. Plant Cell Physiol 51:546–553
Kumar A, Giridhar P (2015) Salicylic acid and methyl jasmonate restore the transcription of caffeine biosynthetic N-methyltransferases from a transcription inhibition noticed during late endosperm maturation in coffee. Plant Gene 4:38–44
Kumar A, Naik GK, Giridhar P (2017) Dataset on exogenous application of salicylic acid and methyl jasmonate and the accumulation of caffeine in young leaf tissues and catabolically inactive endosperms. Data Brief 13:22–27
Kutywayo D, Chemura A, Kusena W, Chidoko P, Mahoya C (2013) The impact of climate change on the potential distribution of agricultural pests: the case of the coffee white stem borer (Monochamus leuconotus P.) in Zimbabwe. PLoS One 8:e73432
Ky CL, Louarn J, Dussert S, Guyot B, Hamon S, Noirot M (2001) Caffeine, trigonelline, chlorogenic acids and sucrose diversity in wild Coffea arabica L. and C. canephora P. accessions. Food Chem 75:223–230
Lashermes P, Couturon E, Moreau N, Paillard M, Louarn J (1996) Inheritance and genetic mapping of self-incompatibility in Coffea canephora Pierre. Theor Appl Genet 93:458–462
Lashermes P, Combes MC, Robert J, Trouslot P, D’Hont A, Anthony F, Charrier A (1999) Molecular characterisation and origin of the Coffea arabica L. genome. Mol Gen Genet 261:259–266
Lashermes P, Andrade AC, Etienne H (2008) Genomics of coffee, one of the world’s largest traded commodities. In: Moore H, Ming R (eds) Genomics of tropical crop plants. Springer, Berlin, pp 203–226
Lashermes P, Hueber Y, Combes M-C, Severac D, Dereeper A (2016) Inter-genomic DNA exchanges and homeologous gene silencing shaped the nascent allopolyploid coffee genome (Coffea arabica L.). G3 Genes Genom Genet 6:2937–2948
Lepelley M, Cheminade G, Tremillon N, Simkin A, Caillet V, McCarthy J (2007) Chlorogenic acid synthesis in coffee: an analysis of CGA content and real-time RT-PCR expression of HCT, HQT, C3H1, and CCoAOMT1 genes during grain development in C. canephora. Plant Sci 172:978–996
Lepelley M, Ben Amor M, Martineau N, Cheminade G, Caillet V, McCarthy J (2012a) Coffee cysteine proteinases and related inhibitors with high expression during grain maturation and germination. BMC Plant Biol 12:31
Lepelley M, Mahesh V, McCarthy J, Rigoreau M, Crouzillat D, Chabrillange N, de Kochko A, Campa C (2012b) Characterization, high-resolution mapping and differential expression of three homologous PAL genes in Coffea canephora Pierre (Rubiaceae). Planta 236:313–326
Leroy T, Ribeyre F, Bertrand B, Charmetant P, Dufour M, Montagnon C, Marraccini P, Pot D (2006) Genetics of coffee quality. Braz J Plant Physiol 18:229–242
Li B, Kadura I, Fu DJ, Watson DE (2004) Genotyping with TaqMAMA. Genomics 83:311–320
Lima EA (2015) Resistência múltipla do cafeeiro Coffea canephora população Conilon a Meloidogyne spp.: mecanismos e possíveis genes envolvidos. PhD dissertation, Federal University of Brasilia, Brazil, p 148
Lima EA, Carneiro FA, Costa TS, Rêgo ECS, Jorge A Jr, Furlanetto C, Marraccini P, Carneiro RMDG, Andrade AC (2014) Molecular characterization of resistance responses of C. canephora ‘Clone 14’ upon infection by Meloidogyne paranaensis. J Nematol 46:194
Lima EA, Furlanetto C, Nicole M, Gomes ACMM, Almeida MRA, Jorge-Júnior A, Correa VR, Salgado SM, Ferrão MAG, Carneiro RMDG (2015) The multi-resistant reaction of drought-tolerant coffee ‘Conilon clone 14’ to Meloidogyne spp. and late hypersensitive-like response in Coffea canephora. Phytopathology 105:805–814
Lin C, Mueller LA, Mc Carthy J, Crouzillat D, Pétiard V, Tanksley S (2005) Coffee and tomato share common gene repertoires as revealed by deep sequencing of seed and cherry transcripts. Theor Appl Genet 112:114–130
Loss-Morais G, Ferreira DCR, Margis R, Alves-Ferreira M, Corrêa RL (2014) Identification of novel and conserved microRNAs in Coffea canephora and Coffea arabica. Genet Mol Biol 37:671–682
Loureiro A, Nicole MR, Várzea V, Moncada P, Bertrand B, Silva MC (2012) Coffee resistance to Colletotrichum kahawae is associated with lignification, accumulation of phenols and cell death at infection sites. Physiol Mol Plant Pathol 77:23–32
Ma C, Burd S, Lers A (2015) miR408 is involved in abiotic stress responses in Arabidopsis. Plant J 84:169–187
Magalhães STV, Fernandes FL, Demuner AJ, Picanço MC, Guedes RNC (2010) Leaf alkaloids, phenolics, and coffee resistance to the leaf miner Leucoptera coffeella (Lepidoptera: Lyonetiidae). J Econ Entomol 103:1438–1443
Magrach A, Ghazoul J (2015) Climate and pest-driven geographic shifts in global coffee production: implications for forest cover, biodiversity and carbon storage. PLoS One 10:e0133071
Maluf MP, da Silva CC, de Oliveira MP, Tavares AG, Silvarolla MB, Filho OG (2009) Altered expression of the caffeine synthase gene in a naturally caffeine-free mutant of Coffea arabica. Genet Mol Biol 32:802–810
Marraccini P, Deshayes A, Pétiard V, Rogers WJ (1999) Molecular cloning of the complete 11S seed storage protein gene of Coffea arabica and promoter analysis in transgenic tobacco plants. Plant Physiol Biochem 37:273–282
Marraccini P, Rogers WJ, Allard C, André ML, Caillet V, Lacoste N, Lausanne F, Michaux S (2001) Molecular and biochemical characterization of endo-β-mannanases from germinating coffee (Coffea arabica) grains. Planta 213:296–308
Marraccini P, Courjault C, Caillet V, Lausanne F, Lepage B, Rogers WJ, Tessereau S, Deshayes A (2003) Rubisco small subunit of Coffea arabica: cDNA sequence, gene cloning and promoter analysis in transgenic tobacco plants. Plant Physiol Biochem 41:17–25
Marraccini P, Rogers WJ, Caillet V, Deshayes A, Granato D, Lausanne F, Lechat S, Pridmore D, Pétiard V (2005) Biochemical and molecular characterization of α-D-galactosidase from coffee beans. Plant Physiol Biochem 43:909–920
Marraccini P, Freire LP, Alves GSC, Vieira NG, Vinecky F, Elbelt S, Ramos HJO, Montagnon C, Vieira LGE, Leroy T, Pot D, Silva VA, Rodrigues GC, Andrade AC (2011) RBCS1 expression in coffee: Coffea orthologs, Coffea arabica homeologs, and expression variability between genotypes and under drought stress. BMC Plant Biol 11:85
Marraccini P, Vinecky F, Alves GSC, Ramos HJO, Elbelt S, Vieira NG, Carneiro FA, Sujii PS, Alekcevetch JC, Silva VA, DaMatta FM, Ferrão MAG, Leroy T, Pot D, Vieira LGE, da Silva FR, Andrade AC (2012) Differentially expressed genes and proteins upon drought acclimation in tolerant and sensitive genotypes of Coffea canephora. J Exp Bot 63:4191–4212
Martinez M, Abraham Z, Gambardella M, Echaide M, Carbonero P, Diaz I (2005) The strawberry gene Cyf1 encodes a phytocystatin with antifungal properties. J Exp Bot 56:1821–1829
Martins LD, Tomaz MA, Lidon FC, DaMatta FM, Ramalho JC (2014) Combined effects of elevated [CO2] and high temperature on leaf mineral balance in Coffea spp. plants. Clim Chang 126:365–379
Martins MQ, Rodrigues WP, Fortunato AS, Leitão AE, Rodrigues AP, Pais IP, Martins LD, Silva MJ, Reboredo FH, Partelli FL, Campostrini E, Tomaz MA, Scotti-Campos P, Ribeiro-Barros AI, Lidon FJC, DaMatta FM, Ramalho JC (2016) Protective response mechanisms to heat stress in interaction with high [CO2] conditions in Coffea spp. Front Plant Sci 7:947
Martins MQ, Fortunato AS, Rodrigues WP, Partelli FL, Campostrini E, Lidon FC, DaMatta FM, Ramalho JC, Ribeiro-Barros AI (2017) Selection and validation of reference genes for accurate RT-qPCR data normalization in Coffea spp. under a climate changes context of interacting elevated [CO2] and temperature. Front Plant Sci 8:307
Matiello JB, de Almeida SR, da Silva MB, Ferreira IB, de Carvalho CHS (2015) Siriema AS1, coffee cultivar with resistance to leaf rust and leaf miner. In: IX Simpósio de Pesquisa dos Cafés do Brasil, Curitiba, Brazil, p 190
Mazzafera P, Eskes AB, Parvais JP, Carvalho A (1990) Male sterility detected in Coffea arabica and C. canephora in Brazil. In: 10st international conference on coffee science (ASIC), Paipa, Colombia, pp 466–473
Menezes-Silva PE, Sanglard LMVP, Ávila RT, Morais LE, Martins SCV, Nobres P, Patreze CM, Ferreira MA, Araújo WL, Fernie AR, DaMatta FM (2017) Photosynthetic and metabolic acclimation to repeated drought events play key roles in drought tolerance in coffee. J Exp Bot 68:4309–4322
Miniussi M, Del Terra L, Savi T, Pallavicini A, Nardini A (2015) Aquaporins in Coffea arabica L.: identification, expression, and impacts on plant water relations and hydraulics. Plant Physiol Biochem 95:92–102
Mizuno K, Kato M, Irino F, Yoneyama N, Fujimura T, Ashihara H (2003a) The first committed step reaction of caffeine biosynthesis: 7-methylxanthosine synthase is closely homologous to caffeine synthases in coffee (Coffea arabica L.). FEBS Lett 547:56–60
Mizuno K, Kuda A, Kato M, Yoneyama N, Tanaka H, Ashihara H, Fujimura T (2003b) Isolation of a new dual-functional caffeine synthase gene encoding an enzyme for the conversion of 7-methylxanthine to caffeine from coffee (Coffea arabica L.). FEBS Lett 534:75–81
Mizuno K, Matsuzaki M, Kanazawa S, Tokiwano T, Yoshizawa Y, Kato M (2014) Conversion of nicotinic acid to trigonelline is catalyzed by N-methyltransferase belonged to motif B′ methyltransferase family in Coffea arabica. Biochem Biophys Res Commun 452:1060–1066
Moat J, Williams J, Baena S, Wilkinson T, Gole TW, Challa ZK, Demissew S, Davis AP (2017) Resilience potential of the Ethiopian coffee sector under climate change. Nat Plants 19:17081
Moat J, Gole TW, Davis AP (2019) Least concern to endangered: applying climate change projections profoundly influences the extinction risk assessment for wild Arabica coffee. Glob Chang Biol 25:390–403
Mofatto LS, Carneiro FA, Vieira NG, Duarte KE, Vidal RO, Alekcevetch JC, Guitton MG, Verdeil J-L, Lapeyre-Montes F, Lartaud M, Leroy T, De Bellis F, Pot D, Rodrigues GC, Carazzolle MF, Pereira GAG, Andrade AC, Marraccini P (2016) Identification of candidate genes for drought tolerance in coffee by high-throughput sequencing in the shoot apex of different Coffea arabica cultivars. BMC Plant Biol 16:94
Moisyadi S, Stiles J (1995) A cDNA encoding a metallothionein I-like protein from coffee leaves (Coffea arabica). Plant Physiol 107:295–296
Mondego JMC, Guerreiro-Filho O, Bengtson MH, Drummond RD, Felix JM, Duarte MP, Ramiro D, Maluf MP, Sogayar MC, Menossi M (2005) Isolation and characterization of Coffea genes induced during coffee leaf miner (Leucoptera coffeella) infestation. Plant Sci 169:351–360
Mondego JMC, Vidal RO, Carazzolle MF, Tokuda EK, Parizzi LP, Costa GGL, Pereira LFP, Andrade AC, Colombo CA, Vieira LGE, Pereira GAG, Brazilian Coffee Genome Project Consortium (2011) An EST-based analysis identifies new genes and reveals distinctive gene expression features of Coffea arabica and Coffea canephora. BMC Plant Biol 11:30
Montoya G, Vuong H, Cristancho M, Moncada P, Yepes M (2007) Sequence analysis from leaves, flowers and fruits of Coffea arabica var. Caturra. In: 21st international conference on coffee science (ASIC), Montpellier, France (PB261), pp 717–721
Moraes MS, Teixeira AL, Ramalho AR, Espíndula MC, Ferrão MAG, Rocha RB (2018) Characterization of gametophytic self-incompatibility of superior clones of Coffea canephora. Genet Mol Res 17:gmr16039876
Morais H, Caramori PH, Koguishi MS, Ribeiro AMA (2008) Escala fenológica detalhada da fase reprodutiva de Coffea arabica. Bragantia 67:257–260
Morgante M, Scalabrin S, Scaglione D, Cattonaro F, Magni F, Jurman I, Cerutti M, Liverani FS, Navarini L, Del Terra L, Pellegrino G, Graziosi G, Vitulo N, Valle G (2015) Progress report on the sequencing and assembly of the allotetraploid Coffea arabica var. Bourbon genome. In: Plant and animal genome XXIII. San Diego, CA (USA). https://pag.confex.com/pag/xxiii/webprogram/Paper16802.html
Nellikunnumal SM, Chandrashekar A (2012) Computational identification of conserved microRNA and their targets in Coffea canephora by EST analysis. Dyn Biochem Process Biotech Mol Biol 6:70–76
Nguyen Dinh S, Sai TZT, Nawaz G, Lee K, Kang H (2016) Abiotic stresses affect differently the intron splicing and expression of chloroplast genes in coffee plants (Coffea arabica) and rice (Oryza sativa). J Plant Physiol 201:85–94
Nic-Can GI, López-Torres A, Barredo-Pool F, Wrobel K, Loyola-Vargas VM, Rojas-Herrera R, De-la-Peña C (2013) New insights into somatic embryogenesis: leafy cotyledon1, baby boom1 and WUSCHEL-related homeobox4 are epigenetically regulated in Coffea canephora. PLoS One 8:e72160
Nobile PM, Quecini V, Bazzo B, Quiterio G, Mazzafera P, Colombo CA (2010) Transcriptional profile of genes involved in the biosynthesis of phytate and ferritin in Coffea. J Agric Food Chem 58:3479–3487
Nobres P, Patreze CM, Waltenberg FP, Correa MF, Tavano ECR, Mendes BMJ, Alves-Ferreira M (2016) Characterization of the promoter of the homeobox gene CaHB12 in Coffea arabica. Trop Plant Biol 9:50–62
Nowak MD, Davis AP, Anthony F, Yoder AD (2011) Expression and trans-specific polymorphism of self-incompatibility RNases in Coffea (Rubiaceae). PLoS One 6:e21019
Ogawa M, Herai Y, Koizumi N, Kusano T, Sano H (2001) 7-methylxanthine methyltransferase of coffee plants gene isolation and enzyme properties. J Biol Chem 276:8213–8218
Omondi CO, Agwanda CO, Gichuru EK (2004) Field expression of resistance to coffee berry disease (CBD) as affected by environmental and host-pathogen factors. In: 20th international conference on coffee science (ASIC), Bangalore, India
Ovalle-Rivera O, Läderach P, Bunn C, Obersteiner M, Schroth G (2015) Projected shifts in Coffea arabica suitability among major global producing regions due to climate change. PLoS One 10:e0124155
Park HC, Kim ML, Kang YH, Jeon JM, Yoo JH, Kim MC, Park CY, Jeong JC, Moon BC, Lee JH, Yoon HW, Lee S-H, Chung WS, Lim CO, Lee SY, Hong JC, Cho MJ (2004) Pathogen-and NaCl-induced expression of the SCaM-4 promoter is mediated in part by a GT-1 box that interacts with a GT-1-like transcription factor. Plant Physiol 135:2150–2161
Pereira LFP, Galvão RM, Kobayashi AK, Cação SMB, Vieira LGE (2005) Ethylene production and acc oxidase gene expression during fruit ripening of Coffea arabica L. Braz J Plant Physiol 17:283–289
Pérez-Pascual D, Jiménez-Guillen D, Villanueva-Alonzo H, Souza-Perera R, Godoy-Hernández G, Zúñiga-Aguilar JJ (2018) Ectopic expression of the Coffea canephora SERK1 homolog-induced differential transcription of genes involved in auxin metabolism and in the developmental control of embryogenesis. Physiol Plant 163:530–551
Perrois C, Strickler SR, Mathieu G, Lepelley M, Bedon L, Michaux S, Husson J, Mueller L, Privat I (2015) Differential regulation of caffeine metabolism in Coffea arabica (Arabica) and Coffea canephora (Robusta). Planta 241:179–191
Petitot A-S, Lecouls AC, Fernandez D (2008) Sub-genomic origin and regulation patterns of a duplicated WRKY gene in the allotetraploid species Coffea arabica. Tree Genet Genomes 3:379–390
Petitot A-S, Barsalobres-Cavallari C, Ramiro D, Albuquerque Freire E, Etienne H, Fernandez D (2013) Promoter analysis of the WRKY transcription factors CaWRKY1a and CaWRKY1b homoeologous genes in coffee (Coffea arabica). Plant Cell Rep 32:1263–1276
Pezzopane JRM, Pedro Júnior MJ, Thomaziello RA, Camargo MBP (2003) Escala para avaliação de estádios fenológicos do cafeeiro Arábica. Bragantia 62:499–505
Pinto RT, Freitas NC, Máximo WPF, Cardoso TB, Prudente DO, Paiva LV (2019) Genome-wide analysis, transcription factor network approach and gene expression profile of GH3 genes over early somatic embryogenesis in Coffea spp. BMC Genomics 20:812
Poncet V, Rondeau M, Tranchant C,·Cayrel A, Hamon S, de Kochko A, Hamon P (2006) SSR mining in coffee tree EST databases: potential use of EST–SSRs as markers for the Coffea genus. Mol Gen Genomics 276:436–449
Pré M, Caillet V, Sobilo J, McCarthy J (2008) Characterization and expression analysis of genes directing galactomannan synthesis in coffee. Ann Bot 102:207–220
Privat I, Foucrier S, Prins A, Epalle T, Eychenne M, Kandalaft L, Caillet V, Lin C, Tanksley S, Foyer C, McCarthy J (2008) Differential regulation of grain sucrose accumulation and metabolism in Coffea arabica (Arabica) and Coffea canephora (Robusta) revealed through gene expression and enzyme activity analysis. New Phytol 178:781–797
Privat I, Bardil A, Gomez AB, Severac D, Dantec C, Fuentes I, Mueller L, Joët T, Pot D, Foucrier S, Dussert S, Leroy T, Journot L, De Kochko A, Campa C, Combes M-C, Lashermes P, Bertrand B (2011) The ‘Puce Cafe’ project: the first 15K coffee microarray, a new tool for discovering candidate genes correlated to agronomic and quality traits. BMC Genomics 12:5
Puranik S, Sahu PP, Srivastava PS, Prasad M (2012) NAC proteins: regulation and role in stress tolerance. Trends Plant Sci 17:369–381
Quintana-Escobar AO, Nic-Can GI, Galaz Avalos RM, Loyola-Vargas VM, Gongora-Castillo E (2019) Transcriptome analysis of the induction of somatic embryogenesis in Coffea canephora and the participation of ARF and Aux/IAA genes. PeerJ 7:e7752
Ramalho JC, Rodrigues AP, Semedo JN, Pais IP, Martins LD, Simões-Costa MC, Leitão AE, Fortunato AS, Batista-Santos P, Palos IM, Tomaz MA, Scotti-Campos P, Lidon FC, DaMatta FM (2013) Sustained photosynthetic performance of Coffea spp. under long-term enhanced [CO2]. PLoS One 8:e82712
Ramalho JC, Rodrigues AP, Lidon FC, Marques LMC, Leitão AE, Fortunato AS, Pais IP, Silva MJ, Scotti-Campos P, Lopes A, Reboredo FH, Ribeiro-Barros AI (2018a) Stress cross-response of the antioxidative system promoted by superimposed drought and cold conditions in Coffea spp. PLoS One 13:e0198694
Ramalho JC, Pais IP, Leitão AE, Guerra M, Reboredo FH, Máguas CM, Carvalho ML, Scotti-Campos P, Ribeiro-Barros AI, Lidon FJC, DaMatta FM (2018b) Can elevated air [CO2] conditions mitigate the predicted warming impact on the quality of coffee bean? Front Plant Sci 9:287
Ramiro D, Jalloul A, Petitot A-S, Grossi de Sá MF, Maluf MP, Fernandez D (2010) Identification of coffee WRKY transcription factor genes and expression profiling in resistance responses to pathogens. Tree Genet Genomes 6:767–781
Rebijith KB, Asokan R, Ranjitha HH, Krishna V, Nirmalbabu K (2013) In silico mining of novel microRNAs from coffee (Coffea arabica) using expressed sequence tags. J Hortic Sci Biotechnol 88:325–337
Reymond P, Weber H, Damond M, Farmer EE (2000) Differential gene expression in response to mechanical wounding and insect feeding in Arabidopsis. Plant Cell 12:707–720
Rodrigues WP, Martins MQ, Fortunato AS, Rodrigues AP, Semedo JN, Simões-Costa MC, Pais IP, Leitão AE, Colwell F, Goulao L, Máguas C, Maia R, Partelli FL, Campostrini E, Scotti-Campos P, Ribeiro-Barros AI, Lidon FC, DaMatta FM, Ramalho JC (2016) Long-term elevated air [CO2] strengthens photosynthetic functioning and mitigates the impact of supra-optimal temperatures in tropical Coffea arabica and C. canephora species. Glob Chang Biol 22:415–431
Rogers WJ, Bézard G, Deshayes A, Meyer I, Pétiard V, Marraccini P (1999a) Biochemical and molecular characterization and expression of the 11S-type storage protein from Coffea arabica endosperm. Plant Physiol Biochem 37:261–272
Rogers WJ, Michaux S, Bastin M, Bucheli P (1999b) Changes to the content of sugars, sugar alcohols, myo-inositol, carboxylic acids, and inorganic anions in developing grains from different varieties of Robusta (Coffea canephora) and Arabica (C. arabica) coffees. Plant Sci 149:115–123
Romero JV, Cortina H (2004) Fecundidad y ciclo de vida de la coffee berry borer Hypothenemus hampei F. (Coleoptera: Curculionidae: Scolytinae) en introducciones silvestres de café. Rev Cenicafé 55:221–231
Romero JV, Cortina H (2007) Tablas de vida de Hypothenemus hampei (Coleoptera: Curculionidae: Scolytinae) sobre tres introducciones de café. Rev Colomb Entomol 33:10–16
Rushton PJ, Somssich IE, Ringler P, Shen QJ (2010) WRKY transcription factors. Trends Plant Sci 15:247–258
Ságio SA, Barreto HG, Lima AA, Moreira RO, Rezende PM, Paiva LV, Chalfun-Junior A (2014) Identification and expression analysis of ethylene biosynthesis and signaling genes provides insights into the early and late coffee cultivars ripening pathway. Planta 239:951–963
Salmona J, Dussert S, Descroix F, de Kochko A, Bertrand B, Joët T (2008) Deciphering transcriptional networks that govern Coffea arabica seed development using combined cDNA array and real-time RT-PCR approaches. Plant Mol Biol 66:105–124
Samson N, Bausher MG, Lee SB, Jansen RK, Daniell H (2007) The complete nucleotide sequence of the coffee (Coffea arabica L.) chloroplast genome: organization and implications for biotechnology and phylogenetic relationships amongst angiosperms. Plant Biotechnol J 5:339–353
Sant’Ana GC, Pereira LFP, Pot D, Ivamoto ST, Domingues DS, Ferreira RV, Pagiatto NF, da Silva BSR, Nogueira LM, Kitzberger CSG, Scholz MBS, de Oliveira FF, Sera GH, Padilha L, Labouisse JP, Guyot R, Charmetant P, Leroy T (2018) Genome-wide association study reveals candidate genes influencing lipids and diterpenes contents in Coffea arabica L. Sci Rep 8:465
Santos AB, Mazzafera P (2012) Dehydrins are highly expressed in water-stressed plants of two coffee species. Trop Plant Biol 5:218–232
Santos GC, Von Pinho EVR, Rosa SDVF (2013) Gene expression of coffee seed oxidation and germination processes during drying. Genet Mol Res 12:6968–6982
Santos JO, Santos MO, Torres LF, Matos CSM, Botelho A, Andrade AC, Carvalho GR, Silva VA (2019) Analysis of APX and CaPYL8A genes expression in Coffea arabica progenies under drought. In: X Simpósio de Pesquisa dos Cafés do Brasil, Vitoria, Brazil
Satyanarayana KV, Kumar V, Chandrashekar A, Ravishankar GA (2005) Isolation of promoter for N-methyltransferase gene associated with caffeine biosynthesis in Coffea canephora. J Biotechnol 119:20–25
Schluter U, Benchabane M, Munger A, Kiggundu A, Vorster J, Goulet MC, Cloutier C, Michaud D (2010) Recombinant protease inhibitors for herbivore pest control: a multitrophic perspective. J Exp Bot 61:4169–4183
Scotti-Campos P, Pais IP, Ribeiro-Barros AI, Martins LD, Tomaz MA, Rodrigues WP, Campostrini E, Semedo JN, Fortunato AS, Martins MQ, Partelli FL, Lidon FC, DaMatta FM, Ramalho JC (2019) Lipid profile adjustments may contribute to warming acclimation and to heat impact mitigation by elevated [CO2] in Coffea spp. Environ Exp Bot 167:103856
Selmar D, Bytof G, Knopp SE, Breitenstein B (2006) Germination of coffee seeds and its significance for coffee quality. Plant Biol 8:260–264
Sera GH, Sera T, Ito DS, Filho CR, Villacorta A, Kanayama FS, Alegre CR, Del Grossi L (2010) Coffee berry borer resistance in coffee genotypes. Braz Arch Biol Technol 53:261–268
Severino FE, Brandalise M, Costa CS, Wilcken SRS, Maluf MP, Gonçalves W, Maia IG (2012) CaPrx, a Coffea arabica gene encoding a putative class III peroxidase induced by root-knot nematode infection. Plant Sci 191-192:35–42
Silva EA, Mazzafera P, Brunini O, Sakai E, Arruda FB, Mattoso LHC, Carvalho CRL, Pires RCM (2005) The influence of water management and environmental conditions on the chemical composition and beverage quality of coffee beans. Braz J Plant Physiol 17:229–238
Silva MC, Várzea V, Guerra-Guimarães L, Azinheira HG, Fernandez D, Petitot A-S, Bertrand B, Lashermes P, Nicole M (2006) Coffee resistance to the main diseases: leaf rust and coffee berry disease. Braz J Plant Physiol 18:119–147
Silva AT, Barduche D, do Livramento KG, Ligterink W, Paiva LV (2014) Characterization of a putative Serk-Like ortholog in embryogenic cell suspension cultures of Coffea arabica L. Plant Mol Biol Rep 32:176–184
Silva AT, Barduche D, do Livramento KG, Paiva LV (2015) A putative BABY BOOM-like gene (CaBBM) is expressed in embryogenic calli and embryogenic cell suspension culture of Coffea arabica L. In Vitro Cell Dev Biol Plant 51:93
Simkin AJ, Qian T, Caillet V, Michoux F, Ben Amor M, Lin C, Tanksley S, McCarthy J (2006) Oleosin gene family of Coffea canephora: quantitative expression analysis of five oleosin genes in developing and germinating coffee grain. J Plant Physiol 163:691–708
Simkin AJ, Moreau H, Kuntz M, Pagny G, Lin C, Tanksley S, McCarthy J (2008) An investigation of carotenoid biosynthesis in Coffea canephora and Coffea arabica. J Plant Physiol 165:1087–1106
Simkin AJ, Kuntz M, Moreau H, McCarthy J (2010) Carotenoid profiling and the expression of carotenoid biosynthetic genes in developing coffee grain. Plant Physiol Biochem 48:434–442
Singh R, Irikura B, Nagai C, Albert HH, Kumagai M, Paull RE, Moore PH, Wang M-L (2011) Characterization of prolyl oligopeptidase genes differentially expressed between two cultivars of Coffea arabica L. Trop Plant Biol 4:203–216
Sreedharan SP, Kumar A, Giridhar P (2018) Primer design and amplification efficiencies are crucial for reliability of quantitative PCR studies of caffeine biosynthetic N-methyltransferases in coffee. 3. Biotech 8:467
Thioune EH, McCarthy J, Gallagher T, Osborne B (2017) A humidity shock leads to rapid, temperature dependent changes in coffee leaf physiology and gene expression. Tree Physiol 37:367–379
Tiski I, Marraccini P, Pot D, Vieira LGE, Pereira LFP (2011) Characterization and expression of two cDNA encoding 3-hydroxy-3-methylglutaryl coenzyme A reductase isoforms in coffee (Coffea arabica L.). OMICS 15:719–727
Toniutti L, Breitler J-C, Etienne H, Campa C, Doulbeau S, Urban L, Lambot C, Pinilla JH, Bertrand B (2017) Influence of environmental conditions and genetic background of Arabica coffee (C. arabica L) on leaf rust (Hemileia vastatrix) pathogenesis. Front. Plant Sci 8:2025
Toniutti L, Bordeaux M, Klein PE, Bertrand B, Montagnon C (2019a) Association study of tree size and male sterility in a F2 Coffea arabica population. In: 27th international conference on coffee science (ASIC), Portland (OR), USA
Toniutti L, Breitler J-C, Guittin C, Doulbeau S, Etienne H, Campa C, Lambot C, Herrera Pinilla JC, Bertrand B (2019b) An altered circadian clock coupled with a higher photosynthesis efficiency could explain the better agronomic performance of a new coffee clone when compared with a standard variety. Int J Mol Sci 20:736
Torres LF, Diniz LEC, Do Livramento KG, Freire LL, Paiva LV (2015) Gene expression and morphological characterization of cell suspensions of Coffea arabica L. cv. Catiguá MG2 in different cultivation stages. Acta Physiol Plant 37:175
Torres LF, Reichel T, Déchamp E, de Aquino SO, Duarte KE, Alves GSC, Silva AT, Cotta MG, Costa TS, Diniz LEC, Breitler J-C, Collin M, Paiva LV, Andrade AC, Etienne H, Marraccini P (2019) Expression of DREB-like genes in Coffea canephora and C. arabica subjected to various types of abiotic stress. Trop Plant Biol 12:98–116
Tran HTM, Lee LS, Furtado A, Smyth H, Henry RJ (2016) Advances in genomics for the improvement of quality in coffee. J Sci Food Agric 96:3300–3312
Tran HTM, Ramaraj T, Furtado A, Lee LS, Henry RJ (2018) Use of a draft genome of coffee (Coffea arabica) to identify SNPs associated with caffeine content. Plant Biotechnol 16:1756–1766
Uefuji H, Ogita S, Yamaguchi Y, Koizumi N, Sano H (2003) Molecular cloning and functional characterization of three distinct N-methyltransferases involved in the caffeine biosynthetic pathway in coffee plants. Plant Physiol 132:372–380
Ülker B, Somssich IE (2004) WRKY transcription factors: from DNA binding towards biological functions. Curr Opin Plant Biol 7:491–498
Urwin P, Green J, Atkinson H (2003) Expression of a plant cystatin confers partial resistance to Globodera, full resistance is achieved by pyramiding a cystatin with natural resistance. Mol Breed 12:263–269
Valadez-González N, Colli-Mull JG, Brito-Argáez L, Muñoz-Sánchez JA, Zúñiga Aguilar JJ, Castaño E, Hernández-Sotomayor SMT (2007) Differential effect of aluminum on DNA synthesis and CDKA activity in two Coffea arabica cell lines. J Plant Growth Regul 26:69–77
Valeriano JC, Lima EA, de Aquino SO, Carneiro FA, Carneiro WWJ, Mattos NG, Carneiro RMDG, Andrade AC (2019) Allele specific expression in Coffea canephora associated with nematoides resistance. In: X Simpósio de Pesquisa dos Cafés do Brasil, Vitoria, Brazil
Van der Vossen HAM (1985) Coffee breeding and selection. In: Clifford MN, Wilson RC (eds) Coffee botany, biochemistry and production of beans and beverages. Croom Helm, London, pp 48–97
van der Vossen HAM, Walyaro DJ (2009) Additional evidence for oligogenic inheritance of durable host resistance to coffee berry disease (Colletotrichum kahawae) in arabica coffee (Coffea arabica L.). Euphytica 165:105–111
Van der Vossen H, Bertrand B, Charrier A (2015) Next generation variety development for sustainable production of arabica coffee (Coffea arabica L.): a review. Euphytica 204:243–256
Vasconcelos EAR, Santana CG, Godoy CV, Seixas CDS, Silva MS, Moreira LRS, Oliveira-Neto OB, Price D, Fitches E, Filho EXF, Mehta A, Gatehouse JA, Grossi-de-Sa MF (2011) A new chitinase-like xylanase inhibitor protein (XIP) from coffee (Coffea arabica) affects soybean Asian rust (Phakopsora pachyrhizi) spore germination. BMC Biotechnol 11:14
Vidal RO, Mondego JMC, Pot D, Ambrósio AB, Andrade AC, Pereira LFP, Colombo CA, Vieira LGE, Carazzolle MF, Pereira GAG (2010) A high-throughput data mining of single nucleotide polymorphisms in Coffea species expressed sequence tags suggests differential homeologous gene expression in the allotetraploid Coffea arabica. Plant Physiol 154:1053–1066
Vieira LGE, Andrade AC, Colombo CA, Moraes AAH, Metha A, Oliveira AC, Labate CA, Marino CL, Monteiro-Vitorello CB, Monte DC, Giglioti E, Kimura ET, Romano E, Kuramae EE, Lemos EGM, Almeida ERP, Jorge EC, Barros EVSA, da Silva FR, Vinecky F, Sawazaki HE, Dorry HFA, Carrer H, Abreu IN, Batista JAN, Teixeira JB, Kitajima JP, Xavier KG, Lima LM, Camargo LEA, Pereira LFP, Coutinho LL, Lemos MVF, Romano MR, Machado MA, Costa MMC, Grossi de Sá MF, Goldman MHS, Ferro MIT, Tinoco MLP, Oliveira MC, Sluys MAV, Shimizu MS, Maluf MP, Eira MTS, Guerreiro Filho O, Arruda P, Mazzafera P, Mariani PDSC, Oliveira RL, Harakava R, Balbao SF, Tsai SM, Mauro SMZ, Santos SN, Siqueira WJ, Costa GGL, Formighieri EF, Carazzolle MF, Pereira GAG (2006) Brazilian coffee genome project: an EST-based genomic resource. Braz J Plant Physiol 18:95–108
Vieira NG, Carneiro FA, Sujii PS, Alekcevetch JC, Freire LP, Vinecky F, Elbelt S, Silva VA, DaMatta FM, Ferrão MAG, Marraccini P, Andrade AC (2013) Different molecular mechanisms account for drought tolerance in Coffea canephora var. Conilon. Trop Plant Biol 6:181–190
Vieira NG, Ferrari IF, Rezende JC, Mayer JLS, Mondego JMC (2019) Homeologous regulation of Frigida-like genes provides insights on reproductive development and somatic embryogenesis in the allotetraploid Coffea arabica. Sci Rep 9:8446
Vinecky F, da Silva FR, Andrade AC (2012) In silico analysis of cDNA libraries SH2 and SH3 for the identification of genes responsive to drought in coffee. Coffee Sci 7:1–19
Vinecky F, Davrieux F, Mera AC, Alves GSC, Lavagnini G, Leroy T, Bonnot F, Rocha OC, Bartholo GF, Guerra AF, Rodrigues GC, Marraccini P, Andrade AC (2017) Controlled irrigation and nitrogen, phosphorous and potassium fertilization affect the biochemical composition and quality of Arabica coffee beans. J Agric Sci 155:902–918
Walyaro DJ (1983) Considerations in breeding for improved yield and quality in arabica coffee (Coffea arabica L.). Thesis, University of Wageningen, Netherlands
Wang J, Tian L, Lee HS, Chen ZJ (2006) Nonadditive regulation of FRI and FLC loci mediates flowering-time variation in Arabidopsis allopolyploids. Genetics 173:965–974
Wang Z, Liu Q, Wang H, Zhang H, Xu X, Li C, Yang C (2016) Comprehensive analysis of trihelix genes and their expression under biotic and abiotic stresses in Populus trichocarpa. Sci Rep 6:36274
Xie F, Xiao P, Chen D, Xu L, Zhang B (2012) miRDeepFinder: a miRNA analysis tool for deep sequencing of plant small RNAs. Plant Mol Biol 80:75–84
Yepes M, Gaitan A, Cristancho MA, Rivera LF, Correa JC, Maldonado CE, Gongora CE, Villegas MA, Posada H, Zimin A, Yorke JA, Aldwinckle H (2016) Building high quality reference genome assemblies using PACBio long reads for the allotetraploid Coffea arabica and its diploid ancestral maternal species Coffea eugenioides. In: Plant and animal genome XXIII. San Diego, CA (USA). https://pag.confex.com/pag/xxiv/webprogram/Paper22250.html
Yuyama PM, Reis Júnior O, Ivamoto ST, Domingues DS, Carazzolle MF, Pereira GAG, Charmetant P, Leroy T, Pereira LFP (2016) Transcriptome analysis in Coffea eugenioides, an Arabica coffee ancestor, reveals differentially expressed genes in leaves and fruits. Mol Gen Genomics 291:323–336
Zheng XQ, Ashihara H (2004) Distribution, biosynthesis and function of purine and pyridine alkaloids in Coffea arabica seedlings. Plant Sci 166:807–813
Zheng XQ, Nagai C, Ashihara H (2004) Pyridine nucleotide cycle and trigonelline (N-methylnicotinic acid) synthesis in developing leaves and fruits of Coffea arabica. Physiol Plant 122:404–411
Zhu A, Goldstein J (1994) Cloning and functional expression of a cDNA encoding coffee bean α-galactosidase. Gene 140:227–231
Zhu C, Ding Y, Liu H (2011) MiR398 and plant stress responses. Physiol Plant 143:1–9
Acknowledgments
I would like to thank B. Bertrand and H. Etienne (UMR IPME, research team “CoffeeAdapt” of UMR IPME, https://umr-ipme.ird.fr/equipes/equipe-coffeeadapt) for their critical reading of the manuscript. This work was supported by the European Union, Program Horizon 2020, call H2020-SFS-2016-2, action RIA, project BREEDCAFS (http://www.breedcafs.eu/), and proposal 727934.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Marraccini, P. (2020). Gene Expression in Coffee. In: Cánovas, F.M., Lüttge, U., Risueño, MC., Pretzsch, H. (eds) Progress in Botany Vol. 82. Progress in Botany, vol 82. Springer, Cham. https://doi.org/10.1007/124_2020_42
Download citation
DOI: https://doi.org/10.1007/124_2020_42
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-68619-2
Online ISBN: 978-3-030-68620-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)