
Abstract
Among neoplasia-associated epigenetic alterations, changes in cellular glycosylation have recently received attention as a key component of hematological malignancy progression. Alterations in glycosylation appear to not only directly impact cell growth and survival, but also alter the adhesion of tumor cells and their interactions with the microenvironment, facilitating cancer-induced immunomodulation and eventual metastasis. Changes in glycosylation arise from altered expression of glycosyltransferases, enzymes that catalyze the transfer of saccharide moieties to a wide range of acceptor substrates, such as proteins, lipids, and other saccharides in the endoplasmic reticulum (ER) and Golgi apparatus. Novel glycan structures in hematological malignancies represent new targets for the diagnosis and treatment of blood diseases. This review summarizes studies of the aberrant expression of glycans commonly found in hematological malignancies and their potential mechanisms and defines the specific roles of glycans as drivers or passengers in the development of hematological malignancies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Carbohydrates, together with lipids, proteins, and nucleic acids, are the four major organic compounds in cells. Each has its own important functions in the development of organisms. Among them, the carbohydrates are not only important energy sources and structural components of cells, but can also be employed to modify lipids and proteins via the covalent addition of monosaccharides or even whole oligosaccharides (glycans). This enzyme-catalyzed process of glycosidic linkage of saccharides to other saccharides, proteins, or lipids is called glycosylation (Pinho and Reis 2015). Glycosylation has a fundamental impact on a diverse range of biological processes. Glycosylated lipids or proteins mediate not only processes such as cell development and differentiation, but also biological events such as transport, signaling, folding, and adhesion. Therefore, it is not hard to imagine that minor alterations in glycosylation will lead to a variety of physiological and pathological changes.
Aberrant alteration of glycosylation is a hallmark of cancer (Abroun et al. 2008; Moremen et al. 2012; Pinho and Reis 2015). It has been well documented that aberrant glycans and glycosylation are associated with malignant transformation and the development of cancers, especially in the early stages of cancer development (Marth and Grewal 2008; Moremen et al. 2012). Well-known proteins involved in tumorigenesis and epithelial mesenchymal transition (EMT), such as p53, IκB kinase subunit β (IKKβ), c-Myc, and Snail are all regulated by glycosylation (Yang et al. 2006; Keiko et al. 2009; Chou et al. 1995; Yoon et al. 2014). In addition, glycans, glycoproteins, and glycolipids can be secreted or can leak into serum when cancer cells undergo necrosis and apoptosis. These tumor-derived molecules detected in serum, such as alpha-fetoprotein (AFP), carcinoembryonic antigen (CEA), human epidermal growth factor receptor 2 (HER2), and prostate specific antigen (PSA), can be used as markers for cancer diagnosis and prognosis (Dhanisha et al. 2018; Chandler et al. 2013; Kirwan et al. 2015; Eichhorn et al. 2005; Reis et al. 2010; Silva 2015). At such, the mechanisms by which these abnormal glycosylation products are generated can be investigated to help to reveal the molecular basis of cancers (Ju and Cummings 2002).
During the past decades, remarkable progress has been achieved in our understanding of the role of glycosylation in hematological malignancies. Here we briefly summarize the recent advances in our knowledge of the relationship between aberrant glycosylation and the development of hematological malignancies.
2 Glycosylation
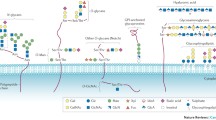
Glycosylation is a complex synergistic process involving enzymes, organelles, and other factors that are necessary for the generation of carbohydrate-associated posttranslational modifications (Stowell et al. 2015). In mammals, the major glycans contain 10 monosaccharide building blocks, glucose (Glc), galactose (Gal), fucose (Fuc), mannose (Man), xylose (Xyl), glucuronic acid (GlcA), iduronic acid (IdoA), β-D-N-acetylglucosamine(GlcNAc), β-D-N-acetylgalactosamine (GalNAc), and 5-N-acetylneuraminic acid (Neu5Ac), all of which can be originally derived from Glc in every cell (Stowell et al. 2015).
As shown in Fig. 1, according to the conserved core structures, glycosylation is roughly divided into N-glycosylation, O-glycosylation, glycosaminoglycan (GAG), glycosphingolipid (GSL), and glycosyl phosphatidylinositol (GPI)-linked proteins (Pang et al. 2018). N-glycosylation is the most common type of glycosylation. The structures of N-glycosylation, that is, N-glycans, which usually have a common conserved pentasaccharide core structure, are linked to asparagine (Asn) residues of proteins, specifically a subset residing in the Asn-X-serine (Ser)/threonine (Thr) (N-X-S/T) motif, in which X can be any amino acid except proline (Pro). The most common abnormal N-glycosylation processes found in hematological malignancies are roughly divided into three categories including sialylation, fucosylation, and bisecting GlcNAc (Pang et al. 2018). O-glycosylation is another common type of glycosylation essential for the biosynthesis of mucins, and it usually occurs on the Ser and Thr residues of glycoproteins. The most common type of protein O-glycosylation is O-GalNAc modification, which involves α linkage of GalNAc to the hydroxyl group of Ser or Thr by an O-glycosidic bond. Other types of covalent modifications of Ser/Thr residues can involve Glc, Fuc, and GlcNAc. Additionally, O-linked glycans usually have much simpler oligosaccharide structures than N-linked glycans (Pang et al. 2018). GAGs are a class of highly sulfated and long unbranched polysaccharides with a repeating disaccharide structure. GAGs can be mainly categorized into four subclasses: heparan sulfate, chondroitin sulfate, dermatan sulfate, and keratin sulfate. GAG chains are usually covalently attached to Ser or Thr residues of proteins. The GAG chains are indispensable functional parts. They are produced through various biosynthetic pathways and are usually highly sulfated, thus having the ability to bind cytokines, chemokines, or growth factors. Through binding, GAG-modified proteins can change cell growth and differentiation, thereby contributing to the regulation of embryogenesis, angiogenesis, and homeostasis (Pang et al. 2018). In addition to N-glycans and O-glycans, GSLs are composed of glycans attached to sphingolipid ceramide (Cer). The core structure of GSLs includes a monosaccharide, usually Glu or Gal, that is directly attached to Cer molecules. The GSL core glycan structure can be extended by other monosaccharides. Since GSLs on the cell membranes are involved in the regulation of retention, quiescence, mobilization, and homing of hematopoietic stem/progenitor cells, they are important for hematological oncogenesis and ontogenesis (Ogretmen and Hannun 2004; Ratajczak and Adamiak 2015).

Glycosylation and hematological malignancies
Glycosylation is usually catalyzed by glycosyltransferases and glycosidases with distinct substrate specificities. Glycosyltransferases are in charge of “adding” sugar blocks, while glycosidases are in charge of “removing” sugar blocks. Abnormalities in glycosyltransferases and glycosidases are closely related to the development of cancers (Pinho and Reis 2015; Stowell et al. 2015). Glycans can be recognized by lectins, such as selectins, galectins, and calreticulin (CALR), which can modulate activity and signaling in cells, and thus contribute to the regulation of cell physiological and pathological processes. Lectins are glycan-binding proteins that are typically highly selective for specific glycan structures. Aberrant glycosylation alters the abundance of ligands of endogenous lectins, and thereby affects multiple cellular mechanisms involving the corresponding glycans (Pinho and Reis 2015; Stowell et al. 2015).
3 Glycosylation in Hematological Malignancies
Glycan aberrations can be found in most hematological malignancies. Increased glycosylation in hematological malignancies is triggered by overexpression of glycoproteins that carry certain specific glycans and altered expression of glycosyltransferases and glycosidases (Marth and Grewal 2008; Pinho and Reis 2015). These glycoproteins are involved in signaling pathways related to cell proliferation, adhesion inhibition and immune escape. Because changes in glycosylation occur in the early stage of cancer development, the detection of tumor associated-glycosylation markers is an effective strategy that can improve the clinical diagnosis and treatments. Here, we will outline and discuss glycosylation abnormalities in different hematological malignancies, including leukemia, lymphoma, myeloma, and other types of malignancies.
4 Glycosylation in Leukemia
Leukemia is a broad term encompassing several hematological malignancies with increased numbers of leukocytes in blood or bone marrow. Once the hematopoietic stem cells in bone marrow transform into leukemic cells, they will grow and survive better than normal cells, and subsequently suppress the development of normal cells. The type of leukemia depends on the type of blood cells involved and the rate of leukemia progression.
4.1 Acute Myeloid Leukemia (AML)
Acute myeloid leukemia (AML) is the most common type of acute leukemia in adults. The survival rates of AML for both adults and children are very poor, with the overall five-year survival rate is 27–65% (Almeida and Ramos 2016). AML is a heterogeneous blood cancer that results from abnormal proliferation of white blood cells that are initiated and maintained by leukemic stem cells (LSCs) featuring aberrations such as translocations of t(6;9)(p22;q34), t(8;21), t(15;17), or inv(16) (Grimwade et al. 1998; Rowley and Potter 1976). In addition, aberrations of chromosomes 5 and 7 or abnormalities of 11q23 in AML are involved in the poor response to chemotherapy (Schoch et al. 2005).
Aberrant glycosylation, such as abnormalities in N-glycosylation, O-glycosylation, and GAG modifications, can occur in AML (Table 1). Aberrant glycosylation is involved in changes in proliferation-related signaling molecules during tumor formation. NOTCH signaling plays crucial roles in the growth of AML cells. The activation of NOTCH signaling induces overexpression of glycosyltransferases including protein O-fucosyltransferase 1 (POFUT1), Fringe (FNG) glycosyltransferase and protein O-glucosyltransferase 1 (POGLUT1, also known as hCLP46), which further increases the glycosylation levels of O-Fuc and O-Glu in NOTCH (Ma et al. 2011; Chu et al. 2013; Yao et al. 2011; Wang et al. 2018). Glycosylation leads to the increased sensitivity of NOTCH to ligands, further enhancing the NOTCH activity in a positive feedback manner (Wang et al. 2018). FMS-related tyrosine kinase 3 (FLT3), a receptor tyrosine kinase that mediates the signaling involved in the proliferation of hematopoietic stem and progenitor cells during development, has been established as a molecular marker on leukemic blasts of patients with AML (Reilly 2003). FLT3/ITD (internal tandem duplications) mutations have been found in approximately 30% of AML patients (Schmidt-Arras et al. 2005). FLT3 needs to undergo posttranslational modification after translation to serve as a functional cell surface receptor. FLT3 is first glycosylated in the ER to form an immature protein, undergoes final glycosylation in the Golgi complex to become a mature receptor, and then translocates to the cell surface (Williams et al. 2012). FLT3/ITD protein exists predominantly in an immature, underglycosylated 130-kDa form in AML, whereas the wild-type FLT3 protein is a mature, completely glycosylated 150-kDa molecule (Schmidt-Arras et al. 2005). Because they induce cell apoptosis through inhibition of glycosylation of FLT3, fluvastatin and 2-Deoxy-d-glucose (2DG) are potential drugs that can prolong the survival of AML patients in the clinic (Williams et al. 2012; Larrue et al. 2015). In addition to fluvastatin and 2DG, an Fc-optimized antibody termed 4G8SDIEM that directly targets FLT3 was developed to treat AML. This type of antibody was designed by defined modifications of the glycosylation pattern or the amino acid sequence of the human immunoglobulin G1 Fc part to enhance its ability to induce cellular cytotoxicity against FLT3-expressing cell lines as well as blasts of AML patients (Hofmann et al. 2012). In addition, several flavonoid derivatives have the potential to be further optimized as FLT3 inhibitors and provide valuable chemical information for the development of new AML drugs (Yen et al. 2021).
Changes in glycosylation of signaling molecules associated with cell migration and bone marrow homing also contribute to the development of AML. CD82 (also known as Kai1) is considered a metastasis suppressor in solid tumors (Dong et al. 1995). The glycosylation level of CD82 affects the molecular structure of N-cadherin by modulating N-glycosylation and the adhesion and trafficking of AML cells, and thus controls the progression of disease (Marjon et al. 2016). Serglycin (SRGN) is the major cell-associated proteoglycan of hematopoietic cells and the level of SRGN in plasma from patients with AML was reported to be significantly higher than that in plasma from patients with acute lymphoblastic leukemia (ALL) or Philadelphia chromosome-negative chronic myeloproliferative leukemia (CML) (Niemann et al. 2007). An abnormal expression level of SRGN can be used as a marker for AML diagnosis (Niemann et al. 2007). A recent study revealed the underlying mechanism of SRGN in AML. The expression of SRGN can be modulated by small nucleolar RNA host gene 3 (SNHG3), a recently identified long noncoding RNA. Increased expression of SRGN stimulates cell proliferation by enhancing Ki67 expression and inhibits cell apoptosis by reducing caspase 3 expression in AML patients (Peng et al. 2020). Galectins belong to a family of lectins which have high affinities for β-galactosides (Ruvolo 2019). Fifteen galectins have been identified and they are involved in cell differentiation, inflammation, adhesion, migration, and apoptosis (Ruvolo 2019). Previous studies have shown that higher galectin-3 (LGALS3) expression in bone marrow is an independent prognostic factor for poor survival in AML patients (Gao et al. 2017; Cheng et al. 2013; Ruvolo 2019). Galectin-3 is related to the regulation of mesenchymal stromal cell homeostasis, and cell localization and cell survival in the leukemia microenvironment niche for AML (Ruvolo et al. 2018). In addition, another member of the galectin family, galectin-9, is believed to be involved in the immune escape of human myeloid leukemia cells (Goncalves Silva et al. 2017). Because of the important role of galectins in the development of AML, the novel galectin inhibitor GCS-100 may be a potential drug for AML therapy (Ruvolo et al. 2016). E-selectin receptor CD162 was reported that it is associated with AML chemo-resistance (Erbani et al. 2020). Actually, the alterations in cell surface glycosylation associated with oncogenesis enhance AML blast binding to E-selectin and enable promotion of pro-survival signaling through AKT/NF-κB pathways. Absence or therapeutic blockade of E-selectin using small molecule mimetic GMI-1271/Uproleselan effectively inhibits this niche-mediated pro-survival signaling and dampens AML blast regeneration (Barbier et al. 2020).
Changes in glycosylation also affect molecules associated with the drug resistance in AML. Cer is an apoptosis inducer in response to chemotherapy in AML (Haimovitz-Friedman et al. 1997; Hannun and Obeid 1997). P-glycoprotein (P-gp) can be targeted to reverse resistance to Cer-induced apoptosis (Turzanski et al. 2005) and has already been used as a classical marker for overcoming multidrug resistance (MDR) in AML (Del Poeta et al. 1996). Specific inhibition of glycosylation with the P-gp inhibitor chlamycin caused a decrease in the activity of glucose ceramide synthase (GCS) (Turzanski et al. 2005). Therefore, it is speculated that the underlying mechanism of MDR in AML is caused by increased enzyme activity of GCS, which is involved in the synthesis of P-gp. Low expression of glycosyltransferase ALG9 is a predicted poor prognosis of AML patients and lncRNAs target ALG9, which plays a post-transcriptional regulatory role providing a novel therapeutic target for AML chemo-resistance (Yu et al. 2020).
In addition, proteomic analysis showed that heterogeneous nuclear ribonucleoprotein H1 (HNRNPH1), an RNA-binding protein, was overexpressed and modified with O-GlcNAc in AML patients with chromosome 11q23 ectopic translocation, although the association between HNRNPH1 and glycosylation modification in AML remains to be further investigated (Balkhi et al. 2006). Although many glycosylation aberrations have been found in AML patients, to the best of our knowledge, glycosylation changes have not been reported in relatively rare subtypes of AML, such as acute megakaryoblastic leukemia, which is associated with a high risk of Down’s syndrome children.
4.2 Chronic Myeloid Leukemia (CML)
CML is caused by a chromosomal translocation between the long arms of chromosomes 9 and 22 that forms the fusion gene Bcr-abl1 (PeterC.Nowell 2002; Stagno et al. 2016). The Bcr-abl1 gene encodes the oncoprotein BCR-ABL1, which has constitutively increased tyrosine kinase activity, resulting in aggression and proliferation advantage of hematopoietic stem cells (Ren 2005; Giallongo et al. 2011; Stella et al. 2013; Manzella et al. 2016; Preyer et al. 2011; Massimino et al. 2014). As shown in Table 1, glycosylation aberrations have been reported in CML.
Currently, most of the studies on glycosylation modification in CML focus on N-glycosylation. Abnormal modification of glycoprotein components on the surface of macromolecules of CML platelets was the first to be reported. Platelets from CML patients show elevated galactosyl and GalNAc modifications of macromolecular surface glycoprotein constituents (Vainer and Bussel 1976). Later, a study showed abnormalities in the terminal sialic acid residue of platelet membrane glycoprotein IIIb and the penultimate Gal/GalNAc residues of GP Ib, IIb, IIIa, and IIIb (Clezardin et al. 1985). Subsequently, leukocyte interferon alpha (IFNα) isolated from CML patients showed various degrees of glycosylation, such as glucosamine and galactosamine modifications (Labdon et al. 1984). At present, sialylation of glycoproteins seems to be a major feature of immature granulocytes in CML. Granulocytes in CML patients exhibit markedly increased sialylation of glycoproteins on the cell surface, which may be due to an increased activity of sialyltransferases and altered activity of other glycosyltransferases and sialidases (Cyopick et al. 1993). Aberrant glycosylation of CML granulocytes may reduce the binding of hematopoietic growth factors, resulting in aberrant differentiation and proliferation of myeloid-lineage cells (Cyopick et al. 1993). These abnormal sialylation events may be caused by overexpression of the sialyltransferases ST3GAL4 and ST6GAL1 (Nasirikenari et al. 2014; Zhou et al. 2017). Recently, it was reported that a synthetic GalNAc analog, peracetyl N-thioglycolyl-d-galactosamine (Ac5GalNTGc) can regulate immune activation in the CML K562 cell line by inhibiting mucin-type O-glycosylation (MTOG) of CD43 (Dwivedi et al. 2018).
In addition, the disruption of the immune system through glycosylation of the NK cell receptor is another potential mechanism underlying the formation of CML. Glycosylation modification of NK cell surface receptors, particularly NKG2D, results in the failure of these structures to induce their normal effect of NK-mediated lysis (Cebo et al. 2006). In addition, β-1,4-mannosyl-glycoprotein 4-β-N-acetylglucosaminyltransferase 3 (MGAT3) is the key enzyme for the biosynthesis of N-glycans. MGAT3 catalyzes the addition of a bisecting GlcNAc residue to the L-Man of the mannosyl core of N-glycans (Taniguchi et al. 1999). The activity of MGAT3 is obviously elevated in patients with CML in blast crisis (Yoshimura et al. 1995). Overexpression of MGAT3 in K562 leukemia cells results in an increase in bisecting GlcNAc modifications and a decrease in external sialic acid modifications, which changes the binding to effector cells, leading to K562 cell resistance to NK cell cytotoxicity (Yoshimura et al. 1996).
Glycosylation modification is not only crucial for the molecules associated with cell proliferation in the development of CML, but also reflects MDR to tyrosine kinase inhibitors (TKIs), such as imatinib, nilotinib, and dasatinib, in the treatment of CML (Stagno et al. 2016). Acquired resistance to TKIs is frequently associated with poor clinical outcomes in CML patients. The most common causes of resistance can be categorized as BCR-ABL1-dependent and BCR-ABL1-independent. BCR-ABL1-dependent resistance is due to point mutations in the BCR-ABL1 kinase domain that interfere with the binding with TKIs, and BCR-ABL1-independent resistance is mediated by activation of alternative survival pathways under the effective inhibition of TKIs (Ma et al. 2014b; Patel et al. 2017). Another potential mechanism of MDR to TKIs may be the changes in the glycosylation of CML cells. Elevated expression of α-2, 3-sialyltransferase 4 (ST3GAL4) was found in CML cells with imatinib resistance (Taniguchi et al. 1999). In addition, CML cells from patients and cell lines with MDR showed overexpression of α-2, 8-polysialytransferase 4 (ST8SIA4) and ST3GAL4, and insufficient expression of α-2, 3-sialyltransferase 1 (ST3GAL1), ST3GAL5 and α-2, 8-polysialytransferase 6 (ST8SIA6) (Zhang et al. 2015; Li et al. 2016). Furthermore, expression of galectin-3, which was mentioned in the AML section, induced by the leukemia microenvironment can promote cell proliferation and MDR to TKI in CML cells by inducing accumulation of the antiapoptotic protein Mcl-1 and suppression of the bovine SERPINA1-fetal bovine serum albumin complex (Yamamoto-Sugitani et al. 2011; Nakayama et al. 2014).
4.3 Acute Lymphoblastic Leukemia (ALL)
ALL is a type of blood and bone marrow cancer caused by the neoplasm of immature lymphoid progenitors (Mullighan 2012). ALL is the most common cancer and the most frequent cause of cancer-related death among children and teenagers (Hunger and Mullighan 2015). Based on the type of lymphoid linage, ALL is divided into different types, B cell type (B-ALL) and T cell type (T-ALL); most childhood cases of ALL are B-ALL. More than 75% of B-ALL cases show aneuploidy or recurring gross chromosomal rearrangements, such as ETV6-RUNX1, TCF3-PBX1, and BCR-ABL1, and other mutations, such as mutations in SH2B3 and CDKN2A/CDKN2B, as we reported before (Cheng et al. 2016). T-ALL cases always exhibit mutations in the oncogenic transcription factors TAL1 and TLX1 (Mullighan 2012; Hunger and Mullighan 2015). In addition to the genetic changes in ALL, the aberrant glycosylation involved in the development of ALL has drawn attention (Table 1). Currently, the types of aberrant glycosylation involved in ALL are O-glycosylation and N-glycosylation, which affect the processes of cell adhesion, proliferation, antiapoptosis, and MDR.
C-type lectins like DC-SIGN and L-SIGN are a diverse group of proteins involved in many human physiological and pathological processes. Aberrant glycosylation of blast cells can alter their interaction with C-type lectins and induce an immunosuppressive response. Recent evidence has shown that leukemic blasts from B-ALL and T-ALL patients have increased binding with C-type lectins thereby affecting their immunological elimination (Gijzen et al. 2008). In addition, increased binding of B-ALL peripheral blood cells to DC-SIGN and L-SIGN is correlated with poor prognosis (Gijzen et al. 2008). Macrophage galactose-type lectin (MGL) is another C-type lectin. A recent study found that MGL can recognize terminal GalNAc-containing structures, such as the specific Tn antigen expressed on T cell leukemia cells (Pirro et al. 2019). Hematopoietic cell L-selectin ligand (HCLL) is modified by N-glycosylation with sialylated and fucosylated structures. These structures act as a complete membrane glycoprotein that is expressed in normal hematopoietic cells. However, HCLL from the blasts of ALL patients shows highly sialofucosylated structures on complex-type N-glycans and exhibits higher activity than HCLL from normal cells (Sackstein and Dimitroff 2000). It was also reported that as a marker of platelet activation, P-selectin may be a new relevant marker, which is easier to be analyzed in clinical practice (Pluchart et al. 2021).
Besides lectin receptors and lectin ligands on ALL blasts, another study found that transferrin receptors (TrfRs) are highly expressed on blasts from T-ALL patients; however, molecular weight of TrfRs was decreased due to the absence of certain types of glycosylation, although the types of glycosylation were not investigated (Petrini et al. 1989). In addition to the aberrant glycosylation found on ALL blasts, abnormal glycosylation was found on red cells from ALL patients as well. Anemia is a major feature in pediatric B-ALL and T-ALL. The glycosylation in red cells from patients is elevated approximately three folds compared with that in red cells from healthy people (Ghosh et al. 2005).
Another ribonucleoprotein, HNRNPH1, which was mentioned in the AML section, has been recently identified in cases of B-ALL (Ohki et al. 2019). Although there is no evidence to show that the glycosylation of HNRNPH1 is aberrant in these B-ALL patients, changes in glycosylation of HNRNPH1 may promote the development of B-ALL given the role of such modification in AML, in which HNRNPH1 is overexpressed and modified with O-GlcNAc in AML patients. Further investigation may be needed to provide important insights into this molecule.
The abnormal glycoprotein antigens CD9, CD43, CD45, and CD95 are all overexpressed in ALL blasts, which is characteristic of ALL (Komada and Sakurai 1994; Blixt et al. 2012; Dorrie et al. 2002). Heavy O-glycosylation of CD43 and CD45 on the cell surface can help the blasts to reduce cell adhesion, prevent inappropriate cell contact, and facilitate T cell leukemia virus type 1 (HTLV-1) particles to assembly into large, highly infectious structures on the surface of T cells (Mazurov et al. 2012). A study showed that the lectin affinity of the carbohydrate moiety of the CD9 antigen in ALL reflects the presence of N-linked oligosaccharide chains having groups with GlcNAc residues, a Man core and a terminal D-Gal, although the detailed effects of aberrant CD9 glycosylation on the development of ALL are still unknown (Komada and Sakurai 1994).
It was reported that 9-O-acetylated sialoglycoprotein is overexpressed in children with ALL, and sialylation deficiency played a prominent role in promoting the survival of lymphoblasts in ALL (Bandyopadhyay et al. 2005; Ghosh et al. 2007). With changes in 9-O-acetylated sialoglycoconjugates (9-OAcSGs), 9-OAcSA-specific IgG2 is incapable to activate Fc-glycosylation-sensitive effector in ALL, which results in escape from host defenses (Bandyopadhyay et al. 2005). Furthermore, 9-O-acetylated sialoglycoprotein also decreases the activity of the apoptotic protein caspase 3, which protects blasts from apoptosis (Ghosh et al. 2007). This may be another reason for the survival of ALL blasts.
Several glycosyltransferases have been reported to be involved in the development of ALL. The neuraminidase NEU3 (a membrane type- and ganglioside-specific sialidase) regulates the levels of gangliosides and Cers, which are associated with proliferation and apoptosis, by regulating PKC/ERK/p38 MAPK signaling (Tringali et al. 2009). The enzyme POGLUT1, also known as hCLP46, is overexpressed in T-ALL (Wang et al. 2010). Its overexpression enhances activation of NOTCH signaling and regulates cell proliferation in a cell type-dependent manner, similar to that observed in AML (Ma et al. 2011; Chu et al. 2013).
Glycosylation is also involved in the MDR of ALL. When B-lineage leukemia cell lines are treated with neuraminidase and mannosidase inhibitors, resistance to IFNγ treatment is reversed. This result demonstrates that high N-linked glycosylation of CD95 is associated with IFNγ resistance in B-ALL (Dorrie et al. 2002). Therefore, glycosyltransferase inhibitors can be used to modulate the sensitivity of B-ALL blasts to IFNγ and induce cell apoptosis by inhibiting the glycosylation of CD95. The expression level of ST6GAL1, a class of sialyltransferases, is positively correlated with increased risk of pediatric ALL (Mondal et al. 2010). Increased expression of ST6GAL1 is associated with MDR in leukemia cells by regulation of P-gp and MDR-associated protein 1 (MRP1) through PI3K/Akt signaling (Ma et al. 2014a). Recently, a new recombinant human lactoferrin carrying humanized glycosylation (rhLf-h-glycan) has been reported to be a potential therapeutic drug for ALL (Nakamura-Bencomo et al. 2021).
4.4 Chronic Lymphocytic Leukemia (CLL)
Chronic lymphocytic leukemia (CLL), the most frequent leukemia in developed countries, is a disease with clonal expansion of mature-appearing lymphocytes in blood, bone marrow, and secondary lymphoid tissues. CLL is one of the most common type of leukemia in adults, especially in older people, with a median diagnosis age of 72 years (Hallek et al. 2018; Scarfo et al. 2016). More than 95% of cases are B cell phenotype (B-CLL), which is characterized by clonal proliferation and accumulation of mature CD19-positive B cells with co-expressing CD5 and CD23, and few cases are T cell phenotype (T-CLL) (Scarfo et al. 2016; Boelens et al. 2009).
Compared with normal circulating cells, CLL cells express high levels of O-GlcNAcylated proteins, such as p53, c-Myc, and Akt (Table 1). High levels of glycosylation of proteins change the intracellular signaling processes in CLL cells (Shi et al. 2010). O-GlcNAcylation can increase the downstream signaling of Toll-like receptors (TLRs) after cytokine stimulation in CLL cells. However, high baseline O-GlcNAc levels lower the response to the stimulation and result in the resistance to TLR agonists, chemotherapeutic agents, B cell receptor (BCR) crosslinking agents, and mitogens. In addition, O-GlcNAc levels are associated with the grades of tumor aggression in CLL. CLL with aggressive clinical behavior showed less O-GlcNAcylation than indolent cells, which showed relatively highly O-GlcNAcylation (Shi et al. 2010). In addition, O-GlcNAcylation is very important for B cell hemostasis and transduction of BCR-mediated activation signals. Lacking of O-GlcNAc transferase enhances apoptosis of mature B cells and perturbs B cell homeostasis, resulting in severe defects in the activation of BCR signaling (Wu et al. 2017).
N-glycosylation aberrations found in CLL are mainly involved in BCR signaling (Krysov et al. 2010). BCR signaling plays the most important physiological and pathological role in B cell survival and proliferation. Aberrant activation of BCR signaling leads to many B cell malignancies including CLL (Burger and Chiorazzi 2013). BCRs comprise surface immunoglobulins (sIgs) and CD79a/b. Surface IgM has an important influence on the clinical behavior of CLL. Compared with the mature complex glycan modifications of the μ-constant region of IgM of normal cells, the μ chain of CLL blasts bears immature mannosylated modifications, which should be restricted to the ER and absent on the cell surface (Krysov et al. 2010; Vuillier 2005). Glycosylation and folding defects of the μ chain and CD79a chain, but not the CD79b chain, have been reported in CLL patients. The defects were associated with low expression levels of BCR, which is a characteristic of B-CLL (Vuillier 2005). Moreover, the BCR of CLL blasts carries high levels of mannosylation in the heavy chain constant region but not variable region (Sedlik et al. 2016).
Aberrant expression of molecules involved in BCR signaling also changes the glycosylation of the BCR and affects the activation of BCR signaling. TCL1 is expressed in approximately 90% of human CLL patients and its overexpression leads to the activation of BCR signaling (Herling et al. 2009; Kriss et al. 2012). Overexpression of TCL1 increases the expression of IgM and alters the N-glycosylation of Igα and Igβ, which contributes to the hyperactivation of BCR signaling in CLL (Kriss et al. 2012).
Lectins are also involved in the development of CLL (Table 1). The lymphocyte homing receptor L-selectin (CD62L) is a crucial molecule mediating the binding of B-CLL cells to high endothelial venule (HEV) walls of lymph nodes (Lafouresse et al. 2015). Lymph nodes are sites of malignant hyperplasia, and their enlargement is related to poor prognosis. Aberrant glycosylation of L-selectin accounts for the aggressive cell growth and MDR of CLL (Lafouresse et al. 2015). Galectin-1 is a β-galactoside-binding lectin and is secreted by myeloid cells to establish the appropriate microenvironment to facilitate CLL blast proliferation and invasiveness by inhibiting T cell-mediated immunity (Croci et al. 2013).
Given the role of aberrant glycosylation in the pathogenesis of CLL, targeting certain glycoproteins is an effective therapeutic strategy. Several drugs including glycol-engineered monoclonal antibodies (obinutuzumab and rituximab), BCR signaling inhibitors (ibrutinib, idelalisib, duvelisib, and acalabrutinib), and a BCL-2 inhibitor (venetoclax) have been used in the clinic (Yelvington 2018; Zinzani et al. 2019; Awan et al. 2019; Brander et al. 2019; Patel et al. 2019). Recently, antibody-drug conjugates (ADCs) that combine the specificity of monoclonal antibodies (mAbs) recognizing the aberrant glycosylation in tumors with anticancer drugs with powerful cytotoxicity have been introduced, and they may be able to target the aberrant glycosylation of the Tn antigen in CLL and thus become a new therapeutic strategy (Aller et al. 1996; Sedlik et al. 2016).
5 Glycosylation in Lymphomas
Lymphomas can be divided into Hodgkin’s lymphoma (HL) and non-Hodgkin’s lymphoma (non-HL). More than 90% of cases are non-HL, which further classified as B cell type, T cell type, and NK cell type (Mugnaini and Ghosh 2016). Diffuse large B cell lymphoma (DLBCL), follicular lymphoma (FL), and Burkitt lymphoma (BL) are all types of non-HL. Less than 10% of lymphoma cases are HL, which is divided into classical HL and non-classical HL (Mugnaini and Ghosh 2016). Here, we summarize current advances in the understanding of aberrant glycosylation in HL and non-HL. In our knowledge, signaling molecules related to abnormal glycosylation including Gg3, Gb3, Ig VH genes, IgG, IgM, sIg, galectin-3, c-Myc, PSGL-1, galectin-1 and mucin1, have been reported in lymphoma progression (Table 2). Aberrant glycosylation is associated with cell adhesion, malignant growth, and the innate immune system.
5.1 Hodgkin’s Lymphoma (HL)
Studies of the role of glycosylation modification in the development of HL are rare. Limited studies have reported that GSLs, such as gangliotriaosylceramide (asialo GM2), sialosyllactosylceramide (GM3), gangliotriaosylceramide (Gg3), and galectin-1, seem to play critical roles in HL progression. Asialo GM2 is a GSL and serves as a marker for cell lines derived from patients with HL, and the aberrant expression of GM2 results from the accumulation of incomplete GSL synthesis (Kniep et al. 1983; Hakomori 1984). A highly specific interaction between Gg3 and GM3 has also been reported in HL cell (Kojima and Hakomori 1989). Galectin-1 is overexpressed in HL and facilitates immune privilege in classical HL. High Galectin-1 is correlated with poor outcome and the event-free survival (Juszczynski et al. 2007). Therefore, galectin-1 reflects tumor burden and can serve as a predictive biomarker for relapsed classical HL (Ouyang et al. 2013; Kamper et al. 2011).
5.2 Non-Hodgkin’s Lymphoma (Non-HL)
Studies have shown that N-glycosylation changes at specific sites in the immunoglobulin variable heavy chain (Ig VH) genes are very common in FL and in a subset of DLBCLs (Zhu et al. 2002; Odabashian et al. 2020). In addition, the variable regions (V regions) of IgM/IgG of primary lymphoma cells are highly modified by mannosylation, and oligomannose residues are located in the antigen-binding site, possibly precluding conventional antigen binding to create a unique microenvironment to support the growth of tumor cells and escape from the BCR-mediated immune system (Coelho et al. 2010; Radcliffe et al. 2007). Primary cutaneous follicle center lymphoma (PCFCL) is a rare mature B cell lymphoma also be demonstrated acquired N-linked glycosylation motifs in the BCRs (Koning et al. 2019). Another antigen-independent BCR activation pathway has also been reported. Lectin can directly bind to the V region, which usually binds with cognate antigen in sIg from FL cells, through N-linked oligomannose glycans within the antigen-binding domain to activate BCR signaling. This antigen-independent binding will provide a constitutive activation signaling to facilitate FL cell survival and proliferation (Amin et al. 2015; Linley et al. 2015).
Galectin-3 is overexpressed in DLBCL, which is the most common type of non-HL (Clark et al. 2012). Galectin-3 is the only antiapoptotic member of the galectin family. It is secreted by DLBCL cells and binds with a subset of highly glycosylated CD45 with N-acetyllactosamine sequences on the cell surface through C2GnT-1 glycosyltransferase. The binding of galectin-3 with CD45 inhibits the phosphatase activity of CD45, leading to resistance to chemotherapeutic agents in DLBCL cells (Clark et al. 2012).
The oncoprotein c-Myc is overexpressed in many tumors including lymphoma. c-Myc can be dynamically modified by either glycosylation or phosphorylation at the Thr58 site (Kamemura et al. 2002; Chou et al. 1995). The Thr58 site is located in the malignant transformation domain of c-Myc, where mutations are frequently found in BL (Chou et al. 1995). It has been reported that increased O-GlcNAc glycosylation of Thr58 in c-Myc can inhibit the activity of c-Myc and decrease transformation of cells (Kamemura et al. 2002; Chou et al. 1995).
In addition, elevated levels of the GSL Gb3 were found in BL and the abnormal elevation was caused by high enzyme activity of UDP-Gal:LacCer α-galactosyltransferase, which is involved in the synthesis of Gb3 (Sugawara et al. 2017; Wiels et al. 1984).
6 Glycosylation in Multiple Myeloma (MM)
MM is a neoplastic disorder characterized by the clonal proliferation of malignant transformed plasma cells derived from B cells (Caraccio et al. 2020). Plasma cell clones expand in the bone marrow and produce excessive monoclonal immunoglobulins. Patients with MM usually suffer from tumor-induced bone pain and fractures, anemia, renal failure, and immunodeficiency. Occasionally, the malignant transformed plasma cells infiltrate multiple organs and produce other symptoms (Caraccio et al. 2020).
As shown in Table 3, in MM cells, glycosaminoglycans such as the secretory-vesicle proteoglycan SRGN and the cell surface proteoglycan syndecan-1 (CD138) are highly expressed. MM cells constitutively secrete SRGN to facilitate cell adhesion to collagen type I by increasing the expression of collagen-degrading enzymes, such as the matrix metalloproteinases MMP-2 and MMP-9 (Skliris et al. 2013). Syndecan-1 is a heparan sulfate proteoglycan, which is expressed on the surface of MM cells and actively sheds from the cell surface. Syndecan-1 regulates apoptosis and inhibits the growth of MM cells, resulting in reduced osteoclastogenesis and enhanced osteoblastogenesis (Dhodapkar et al. 1998). Furthermore, syndecan-1 promotes activation of PI3K/PKB and RAS/MAPK signaling to enhance cell survival and proliferation in MM cells by binding to hepatocyte growth factor (HGF) (Derksen et al. 2002). Therefore, glycosaminoglycans play crucial roles in MM cell survival, proliferation, and metastasis.
In addition to glycosaminoglycans, altered glycosyltransferases are associated with malignant transformation and metastasis in MM progression. It has been reported that sialyltransferase ST3 β-galactoside α2,3-sialyltransferase 6 (ST3GAL6) is highly expressed in MM cells and patients. It is a key regulator of selectin ligand synthesis and the expression is positively correlated with the homing and engraftment of MM cells (Glavey et al. 2014). The activity of MGAT3, which is elevated in CML patients, was also elevated in patients with MM (Yoshimura et al. 1995).
Lectins are also involved in the pathogenesis of MM (Table 3). Galectin-1 is overexpressed in MM cells and promotes cell survival. Galectin-1 also activates ERK signaling and contributes to the invasive ability in CD45RA negative but not CD45RA positive MM cells by affecting their adhesion to the extracellular matrix and cellular aggregation (Abroun et al. 2008). Galectin-9 has an antiproliferative effect on MM cell lines and patient-derived myeloma cells by inhibiting the JNK and p38 MAPK signaling pathways. Therefore, galectin-9 can be used as a new therapeutic option to treat MM (Kobayashi et al. 2010). P-selectin glycoprotein ligand 1 (PSGL-1, CD162), the major P-selectin ligand, is constantly expressed in MM cells and a marker of lymphoma cells with plasmacytoid differentiation (Florena et al. 2005; Davenpeck et al. 2000). Furthermore, PSGL-1 can be used as a therapeutic target for treatment of MM. Immunotherapy with an anti-PSGL-1 mAb induced complement-mediated lysis of MM cells in vivo (Tripodo et al. 2009).
7 Glycosylation in Other Blood Cancers
As shown in Table 4, aberrant glycosylation alterations are involved in the development of other blood cancers.
7.1 Glycosylation in Myeloproliferative Neoplasms (MPNs)
MPN are a group of clonal hematopoietic diseases involving excessive bone marrow cell production, comprising essential thrombocytosis (ET), polycythemia vera (PV), and primary myelofibrosis (PMF). Most MPN patients show somatically acquired mutations on Jak2, Calf, and Mpl genes, which result in hyperactivation of JAK-STAT signaling (Nangalia and Green 2017). An immature and highly mannosylated form of thrombopoietin (TPO) receptor (MPL/TpoR), which is due to mutation of calreticulin (CALR), has been reported (Pang et al. 2018; Chachoua et al. 2016). The pathogenic CALR mutation interacts with extracellular N-glycosylation residues of MPL through a C-terminal glycan-binding site, leading to TPO-independent activation of MPL and downstream JAK2-STAT5 activation, promoting cell growth and resulting in the development of MPN (Pang et al. 2018; Araki et al. 2016; Elf et al. 2016). The sialylated derivatives of the glycan structure β4-N-acetyllactosamine expression is significantly increased in the platelets isolated from MPNs (Di Buduo et al. 2021). Up to now, there are some clinical drugs for the treatment of MPN, such as ruxolitinib and hydroxyurea; however, there have been no reports of the drug targeting aberrant glycosylation occurred in MPN, since there are very few studies focusing on the role of glycosylation changes in the pathogenesis of MPN.
7.2 Glycosylation in Myelodysplastic Syndrome/Myeloproliferative Neoplasms (MDS/MPNs)
MDS/MPNs are rare and distinct group of myeloid neoplasms with overlapping clinical, laboratory, and morphologic features (Tanaka and Bejar 2019). There are three different MDS/MPNs overlap conditions based on the clinical features, including chronic myelomonocytic leukemia (CMML), atypical chronic myeloid leukemia (aCML), and juvenile myelomonocytic leukemia (JMML) (Tanaka and Bejar 2019). Unlike classical CML, which is driven by the BCR-ABL1 fusion gene, aCML is a BCR-ABL1 negative MDS/MPN, that is characterized by leukocytosis, granulocytic dysplasia, and poor outcomes (Sadigh et al. 2020). At present, the only glycosylation modifications known to be involved in the development of aCML are modifications affecting threonine (Thr) linked O-glycosylation at the T618I and T640N sites of colony-stimulating factor-3 receptor (CSF3R). These aberrations prevent the O-glycosylation of CRF3R and increased receptor dimerization, resulting in ligand-independent activation of CRF3R downstream signaling pathways, such as the JAK and SRC kinase signaling pathways, in aCML patients (Fleischman et al. 2013; Maxson et al. 2014, 2016). To the best of our knowledge, there is no report on the aberrant glycosylation in CMML and JMML.
7.3 Glycosylation in Chronic Neutrophilic Leukemia (CNL)
CNL displays very similar clinical and hematological characteristics to aCML. CNL is diagnosed based on the expansion of neutrophils in both bone marrow and blood, while aCML is characterized by granulocytic dysplasia and an increased number of neutrophil precursors (Maxson et al. 2013). The CSF3R T618I mutation, which was mentioned above, is also a hallmark of CNL and leads to ligand-independent activation due to loss of O-glycosylation of the receptor (Maxson et al. 2014). A recent study demonstrated that the CSF3R T618I mutation produces a protein that can still be glycosylated but that undergoes enhanced spontaneous internalization and degradation which results in a marked decrease in its surface expression (Price et al. 2020). Thus, we can speculate that the underlying mechanism of CSF3R T618I mutation in CNL may be the induction of oncogenic signals through aberrant trafficking and constitutive phosphorylation of the O-glycosylated receptor. In addition, mutation on N610 site of CSF3R was reported. This mutation prevented membrane-proximal N-glycosylation of CSF3R, thereby driving the ligand-independent cellular expansion (Spiciarich et al. 2018). The underlying mechanism is still unknown and needs to be further explored.
8 Conclusion and Perspectives
Glycosylation is one of the most vulnerable cellular processes. Any small pathologic change or metabolic stress will affect glycosylation and result in aberrant glycan and glycoprotein synthesis. Overexpression of glycoproteins, altered expression of sugar donors, and altered enzyme activity of glycosyltransferases and glycosidases are key mechanisms underlying the synthesis of aberrant glycans and glycoprotein forms in the development of hematological malignancies. As summarized in Tables 1, 2, 3, and 4, various glycosylation abnormalities are often found in hematological malignancies.
Although some aberrant glycosylation corresponds to different hematological malignancies, it is worth noting that aberrant glycosylation in the same protein could be involved in different hematological malignancies. For example, the abnormalities of O-glycosylation of NOTCH occur in both AML and ALL (Ma et al. 2011; Yao et al. 2011; Chu et al. 2013; Wang et al. 2018). Another example, the aberrant O-glycosylation of CD45 promotes cell adhesion in ALL, but the N-glycosylation mutation of CD45 occurred in non-HL and plays an important role in drug resistance (Clark et al. 2012; Mazurov et al. 2012). Therefore, microheterogeneity and structural complexity of glycans pose significant analytical challenges. As detection and analysis methods become more accurate, it will be necessary to deeply investigate the glycosylation changes in different types of hematological malignancies to discover aberrant glycosylation patterns and explore the underlying mechanisms. This knowledge will expand the utility of aberrant glycosylation as diagnostic markers and therapeutic targets for hematological malignancies.
Abbreviations
- 2DG:
-
2-Deoxy-d glucose
- 9-OAcSGs:
-
9-O-acetylated sialoglycoconjugates
- Ac5GalNTGc:
-
Peracetyl N-thioglycolyl-d-galactosamine
- aCML:
-
Atypical chronic myeloid leukemia
- ADC:
-
Antibody-drug conjugate
- AFP:
-
α-Fetoprotein
- ALG9:
-
α-1,2-Mannosyltransferase
- ALL:
-
Acute lymphoblastic leukemia
- ALL:
-
Acute lymphoblastic leukemia
- AML:
-
Acute myeloid leukemia
- asialo GM2:
-
Gangliotriaosylceramide
- Asn:
-
Asparagine
- B-ALL:
-
B cell type acute lymphoblastic leukemia
- B-CLL:
-
B cell phenotype chronic lymphocytic leukemia
- BCR:
-
B cell antigen receptor
- BL:
-
Burkitt lymphoma
- CALR:
-
Calreticulin
- CD138:
-
Cell surface proteoglycan syndecan-1
- CD62L:
-
Lymphocyte homing receptor L-selectin
- CD82 :
-
Kai1
- CEA:
-
Carcinoembryonic antigen
- Cer:
-
Ceramide
- CLL:
-
Chronic lymphocytic leukemia
- CML:
-
Chronic myeloid leukemia
- CMML:
-
Chronic myelomonocytic leukemia
- CNL:
-
Chronic neutrophilic leukemia
- CSF3R:
-
Colony-stimulating factor-3 receptor
- DLBCL:
-
Clinical common large B cell lymphoma
- EMT:
-
Epithelial mesenchymal transition
- ET:
-
Essential thrombocytosis
- FL:
-
Follicular lymphoma
- FLT3:
-
FMS-related tyrosine kinase 3
- FNG:
-
Fringe glycosyltransferase
- Fuc:
-
Fucose
- GAG:
-
Glycosaminoglycan
- Gal:
-
Galactose
- GalNAc:
-
β-D-N-acetylgalactosamine
- GCS:
-
Glucose ceramide synthase
- Gg3:
-
Gangliotriaosylceramide
- Glc:
-
Glucose
- GlcA:
-
Glucuronic acid
- GlcNAc:
-
β-D-N-acetylglucosamine
- GM3:
-
Sialosyllactosylceramide
- GPI:
-
Glycosyl phosphatidylinositol
- GSL:
-
Glycosphingolipid
- HCLL:
-
Hematopoietic cell L-selectin ligand
- HER2:
-
Human epidermal growth factor receptor 2
- HEV:
-
High endothelial venule
- HGF:
-
Hepatocyte growth factor
- HL:
-
Hodgkin’s lymphoma
- HNRNPH1:
-
Heterogeneous nuclear ribonucleoprotein H1
- HTLV-1:
-
T cell leukemia virus type 1
- IdoA:
-
Iduronic acid
- IFNα:
-
Interferon alpha
- Ig VH:
-
Immunoglobulin variable heavy chain genes
- IKKβ:
-
IκB kinase subunit β
- ITD:
-
Internal tandem duplications
- JMML:
-
Juvenile myelomonocytic leukemia
- LGALS3:
-
Galectin-3
- LSCs:
-
Leukemic stem cells
- mAb:
-
Monoclonal antibody
- Man:
-
Mannose
- MDR:
-
Multidrug resistance
- MGAT3:
-
Mannosyl-glycoprotein 4-β-N-acetylglucosaminyltransferase 3
- MGL:
-
Macrophage galactose-type lectin
- MM:
-
Multiple myeloma
- MPL/TpoR:
-
TPO receptor
- MPN:
-
Myeloproliferative neoplasm
- MRP1:
-
MDR-associated protein 1
- MTOG:
-
Mucin-type O-glycosylation
- NEU3 :
-
Membrane type- and ganglioside-specific sialidase
- Neu5Ac:
-
5-N-acetylneuraminic acid
- non-HL:
-
Non-Hodgkin’s lymphoma
- Pgp:
-
P-glycoprotein
- PMF:
-
Primary myelofibrosis
- POFUT1:
-
O-fucosyltransferase 1
- POGLUT1/Hclp46:
-
O-glucosyltransferase 1
- PSA:
-
Prostate specific antigen
- PSGL1/CD162:
-
P-selectin glycoprotein ligand 1
- PV:
-
Polycythemia vera
- Ser:
-
Serine
- sIgs:
-
Surface immunoglobulins
- SNHG3:
-
Small nucleolar RNA host gene 3
- SRGN:
-
Serglycin
- ST3GAL1/4/5/6:
-
α-2,3-Sialyltransferase 1/4/5/6
- ST8SIA4/6:
-
α-2,8-Polysialytransferase 4/6
- T-ALL:
-
T cell type acute lymphoblastic leukemia
- T-CLL:
-
T cell phenotype chronic lymphocytic leukemia
- Thr:
-
Threonine
- TKIs:
-
Tyrosine kinase inhibitors
- TLRs:
-
Toll-like receptors
- TPO:
-
Thrombopoietin
- TrfRs:
-
Transferrin receptors
- V regions:
-
Variable regions
- Xyl:
-
Xylose
References
Abroun S, Otsuyama K, Shamsasenjan K, Islam A, Amin J, Iqbal MS, Gondo T, Asaoku H, Kawano MM (2008) Galectin-1 supports the survival of CD45RA(−) primary myeloma cells in vitro. Br J Haematol 142(5):754–765
Aller CT, Kucuk O, Springer GF, Gilman-Sachs A (1996) Flow cytometric analysis of T and Tn epitopes on chronic lymphocytic leukemia cells. Am J Hematol 52(1):29–38
Almeida AM, Ramos F (2016) Acute myeloid leukemia in the older adults. Leuk Res Rep 6:1–7
Amin R, Mourcin F, Uhel F, Pangault C, Ruminy P, Dupre L, Guirriec M, Marchand T, Fest T, Lamy T, Tarte K (2015) DC-SIGN-expressing macrophages trigger activation of mannosylated IgM B-cell receptor in follicular lymphoma. Blood 126(16):1911–1920
Araki M, Yang Y, Masubuchi N, Hironaka Y, Takei H, Morishita S, Mizukami Y, Kan S, Shirane S, Edahiro Y, Sunami Y, Ohsaka A, Komatsu N (2016) Activation of the thrombopoietin receptor by mutant calreticulin in CALR-mutant myeloproliferative neoplasms. Blood 127(10):1307–1316
Awan FT, Schuh A, Brown JR, Furman RR, Pagel JM, Hillmen P, Stephens DM, Woyach J, Bibikova E, Charuworn P, Frigault MM, Hamdy A, Izumi R, Linghu B, Patel P, Wang MH, Byrd JC (2019) Acalabrutinib monotherapy in patients with chronic lymphocytic leukemia who are intolerant to ibrutinib. Blood Adv 3(9):1553–1562
Balkhi MY, Trivedi AK, Geletu M, Christopeit M, Bohlander SK, Behre HM, Behre G (2006) Proteomics of acute myeloid leukaemia: cytogenetic risk groups differ specifically in their proteome, interactome and post-translational protein modifications. Oncogene 25(53):7041–7058
Bandyopadhyay S, Bhattacharyya A, Mallick A, Sen AK, Tripathi G, Das T, Sa G, Bhattacharya DK, Mandal C (2005) Over-expressed IgG2 antibodies against O-acetylated sialoglycoconjugates incapable of proper effector functioning in childhood acute lymphoblastic leukemia. Int Immunol 17(2):177–191
Barbier V, Erbani J, Fiveash C, Davies JM, Tay J, Tallack MR, Lowe J, Magnani JL, Pattabiraman DR, Perkins AC, Lisle J, Rasko JEJ, Levesque JP, Winkler IG (2020) Endothelial E-selectin inhibition improves acute myeloid leukaemia therapy by disrupting vascular niche-mediated chemoresistance. Nat Commun 11(1):2042
Blixt O, Lavrova OI, Mazurov DV, Clo E, Kracun SK, Bovin NV, Filatov AV (2012) Analysis of Tn antigenicity with a panel of new IgM and IgG1 monoclonal antibodies raised against leukemic cells. Glycobiology 22(4):529–542
Boelens J, Lust S, Vanhoecke B, Offner F (2009) Chronic lymphocytic leukaemia. Anticancer Res 29(2):605–615
Brander D, Islam P, Barrientos JC (2019) Tailored treatment strategies for chronic lymphocytic leukemia in a rapidly changing era. Am Soc Clin Oncol Educ Book 39:487–498
Burger JA, Chiorazzi N (2013) B cell receptor signaling in chronic lymphocytic leukemia. Trends Immunol 34(12):592–601
Caraccio C, Krishna S, Phillips DJ, Schurch CM (2020) Bispecific antibodies for multiple myeloma: a review of targets, drugs, clinical trials, and future directions. Front Immunol 11:501
Cebo C, Da Rocha S, Wittnebel S, Turhan AG, Abdelali J, Caillat-Zucman S, Bourhis JH, Chouaib S, Caignard A (2006) The decreased susceptibility of Bcr/Abl targets to NK cell-mediated lysis in response to imatinib mesylate involves modulation of NKG2D ligands, GM1 expression, and synapse formation. J Immunol 176(2):864–872
Chachoua I, Pecquet C, El-Khoury M, Nivarthi H, Albu RI, Marty C, Gryshkova V, Defour JP, Vertenoeil G, Ngo A, Koay A, Raslova H, Courtoy PJ, Choong ML, Plo I, Vainchenker W, Kralovics R, Constantinescu SN (2016) Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood 127(10):1325–1335
Chandler KB, Pompach P, Goldman R, Edwards N (2013) Exploring site-specific N-glycosylation microheterogeneity of haptoglobin using glycopeptide CID tandem mass spectra and glycan database search. J Proteome Res 12(8):3652–3666
Cheng CL, Hou HA, Lee MC, Liu CY, Jhuang JY, Lai YJ, Lin CW, Chen HY, Liu FT, Chou WC, Chen CY, Tang JL, Yao M, Huang SY, Ko BS, Wu SJ, Tsay W, Tien HF (2013) Higher bone marrow LGALS3 expression is an independent unfavorable prognostic factor for overall survival in patients with acute myeloid leukemia. Blood 121(16):3172–3180
Cheng Y, Chikwava K, Wu C, Zhang H, Bhagat A, Pei D, Choi JK, Tong W (2016) LNK/SH2B3 regulates IL-7 receptor signaling in normal and malignant B-progenitors. J Clin Invest 126(4):1267–1281
Chou TY, Dang CV, Hart GW (1995) Glycosylation of the c-Myc transactivation domain. Proc Natl Acad Sci U S A 92(10):4417–4421
Chu Q, Liu L, Wang W (2013) Overexpression of hCLP46 enhances Notch activation and regulates cell proliferation in a cell type-dependent manner. Cell Prolif 46(3):254–262
Clark MC, Pang M, Hsu DK, Liu FT, de Vos S, Gascoyne RD, Said J, Baum LG (2012) Galectin-3 binds to CD45 on diffuse large B-cell lymphoma cells to regulate susceptibility to cell death. Blood 120(23):4635–4644
Clezardin P, McGregor JL, Dechavanne M, Clemetson KJ (1985) Platelet membrane glycoprotein abnormalities in patients with myeloproliferative disorders and secondary thrombocytosis. Br J Haematol 60(2):331–344
Coelho V, Krysov S, Ghaemmaghami AM, Emara M, Potter KN, Johnson P, Packham G, Martinez-Pomares L, Stevenson FK (2010) Glycosylation of surface Ig creates a functional bridge between human follicular lymphoma and microenvironmental lectins. Proc Natl Acad Sci U S A 107(43):18587–18592
Croci DO, Morande PE, Dergan-Dylon S, Borge M, Toscano MA, Stupirski JC, Bezares RF, Avalos JS, Narbaitz M, Gamberale R, Rabinovich GA, Giordano M (2013) Nurse-like cells control the activity of chronic lymphocytic leukemia B cells via galectin-1. Leukemia 27(6):1413–1416
Cyopick P, Culliton R, Brockhausen I, Sutherland DR, Mills GB, Baker M (1993) Role of aberrant sialylation of chronic myeloid leukemia granulocytes on binding and signal transduction by chemotactic peptides and colony stimulating factors. Leuk Lymphoma 11(1–2):79–90
Davenpeck KL, Brummet ME, Hudson SA, Mayer RJ, Bochner BS (2000) Activation of human leukocytes reduces surface P-selectin glycoprotein ligand-1 (PSGL-1, CD162) and adhesion to P-selectin in vitro. J Immunol 165(5):2764–2772
Del Poeta G, Stasi R, Aronica G, Venditti A, Cox MC, Bruno A, Buccisano F, Masi M, Tribalto M, Amadori S, Papa G (1996) Clinical relevance of P-glycoprotein expression in de novo acute myeloid leukemia. Blood 87(5):1997–2004
Derksen PWKR, Evers LM, Van Oers MH, Spaargaren M, Pals ST (2002) Cell surface proteoglycan syndecan-1 mediates hepatocyte growth factor binding and promotes Met signaling in multiple myeloma. Blood 99:1405–1410
Dhanisha SS, Guruvayoorappan C, Drishya S, Abeesh P (2018) Mucins: structural diversity, biosynthesis, its role in pathogenesis and as possible therapeutic targets. Crit Rev Oncol Hematol 122:98–122
Dhodapkar MVAE, Theus A, Lacy M, Langford JK, Barlogie B et al (1998) Syndecan-1 is a multifunctional regulator of myeloma pathobiology: control of tumor cell survival, growth, and bone cell differentiation. Blood 91:2679–2688
Di Buduo CA, Giannini S, Abbonante V, Rosti V, Hoffmeister K, Balduini A (2021) Increased beta4GALT1 expression associates with platelet surface galactosylation and thrombopoietin plasma levels in MPNs. Blood 137(15):2085–2089
Dong JT, Lamb PW, Rinker-Schaeffer CW, Vukanovic J, Ichikawa T, Isaacs JT, Barrett JC (1995) KAI1, a metastasis suppressor gene for prostate cancer on human chromosome 11p11.2. Science 268(5212):884–886
Dorrie J, Sapala K, Zunino SJ (2002) Interferon-gamma increases the expression of glycosylated CD95 in B-leukemic cells: an inducible model to study the role of glycosylation in CD95-signalling and trafficking. Cytokine 18(2):98–107
Dwivedi V, Saini P, Tasneem A, Agarwal K, Sampathkumar SG (2018) Differential inhibition of mucin-type O-glycosylation (MTOG) induced by peracetyl N-thioglycolyl-d-galactosamine (Ac5GalNTGc) in myeloid cells. Biochem Biophys Res Commun 506(1):60–65
Eichhorn SJ, Young RJ, Davies GR (2005) Modeling crystal and molecular deformation in regenerated cellulose fibers. Biomacromolecules 6(1):507–513
Elf S, Abdelfattah NS, Chen E, Perales-Paton J, Rosen EA, Ko A, Peisker F, Florescu N, Giannini S, Wolach O, Morgan EA, Tothova Z, Losman JA, Schneider RK, Al-Shahrour F, Mullally A (2016) Mutant calreticulin requires both its mutant C-terminus and the thrombopoietin receptor for oncogenic transformation. Cancer Discov 6(4):368–381
Erbani J, Tay J, Barbier V, Levesque JP, Winkler IG (2020) Acute myeloid leukemia chemo-resistance is mediated by E-selectin receptor CD162 in bone marrow niches. Front Cell Dev Biol 8:668
Fleischman AG, Maxson JE, Luty SB, Agarwal A, Royer LR, Abel ML, MacManiman JD, Loriaux MM, Druker BJ, Tyner JW (2013) The CSF3R T618I mutation causes a lethal neutrophilic neoplasia in mice that is responsive to therapeutic JAK inhibition. Blood 122(22):3628–3631
Florena AM, Tripodo C, Miceli L, Ingrao S, Porcasi R, Franco V (2005) Identification of CD162 in plasma-cell dyscrasia. Lancet Oncol 6(8):632
Gao N, Yu WZ, Guo NJ, Wang XX, Sun JR (2017) Clinical significance of galectin-3 in patients with adult acute myeloid leukemia: a retrospective cohort study with long-term follow-up and formulation of risk scoring system. Leuk Lymphoma 58(6):1394–1402
Ghosh S, Bandyopadhyay S, Bhattacharya DK, Mandal C (2005) Altered erythrocyte membrane characteristics during anemia in childhood acute lymphoblastic leukemia. Ann Hematol 84(2):76–84
Ghosh S, Bandyopadhyay S, Mukherjee K, Mallick A, Pal S, Mandal C, Bhattacharya DK, Mandal C (2007) O-acetylation of sialic acids is required for the survival of lymphoblasts in childhood acute lymphoblastic leukemia (ALL). Glycoconj J 24(1):17–24
Giallongo C, Tibullo D, La Cava P, Branca A, Parrinello N, Spina P, Stagno F, Conticello C, Chiarenza A, Vigneri P, Palumbo GA, Di Raimondo F (2011) BRIT1/MCPH1 expression in chronic myeloid leukemia and its regulation of the G2/M checkpoint. Acta Haematol 126(4):205–210
Gijzen K, Raymakers RA, Broers KM, Figdor CG, Torensma R (2008) Interaction of acute lymphopblastic leukemia cells with C-type lectins DC-SIGN and L-SIGN. Exp Hematol 36(7):860–870
Glavey SV, Manier S, Natoni A, Sacco A, Moschetta M, Reagan MR, Murillo LS, Sahin I, Wu P, Mishima Y, Zhang Y, Zhang W, Zhang Y, Morgan G, Joshi L, Roccaro AM, Ghobrial IM, O’Dwyer ME (2014) The sialyltransferase ST3GAL6 influences homing and survival in multiple myeloma. Blood 124(11):1765–1776
Goncalves Silva I, Yasinska IM, Sakhnevych SS, Fiedler W, Wellbrock J, Bardelli M, Varani L, Hussain R, Siligardi G, Ceccone G, Berger SM, Ushkaryov YA, Gibbs BF, Fasler-Kan E, Sumbayev VV (2017) The Tim-3-galectin-9 secretory pathway is involved in the immune escape of human acute myeloid leukemia cells. EBioMedicine 22:44–57
Grimwade D, ., Walker H, ., Oliver F, ., Wheatley K, ., Harrison C, ., Harrison G, ., Rees J, ., Hann I, ., Stevens R, ., Burnett A, . (1998) The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council adult and Children's Leukaemia working parties. Blood 92 (7):2322–2333
Haimovitz-Friedman A, Kolesnick RN, Fuks Z (1997) Ceramide signaling in apoptosis. Br Med Bull 53(3):539–553
Hakomori S-I (1984) Glycosphingolipids as differentiation-dependent, tumor-associated markers and as regulators of cell proliferation. Trends Biochem Sci 9(10):7
Hallek M, Shanafelt TD, Eichhorst B (2018) Chronic lymphocytic leukaemia. Lancet 391(10129):1524–1537
Hannun YA, Obeid LM (1997) Mechanisms of ceramide-mediated apoptosis. Adv Exp Med Biol 407:145–149
Herling M, Patel KA, Weit N, Lilienthal N, Hallek M, Keating MJ, Jones D (2009) High TCL1 levels are a marker of B-cell receptor pathway responsiveness and adverse outcome in chronic lymphocytic leukemia. Blood 114(21):4675–4686
Hofmann M, Grosse-Hovest L, Nubling T, Pyz E, Bamberg ML, Aulwurm S, Buhring HJ, Schwartz K, Haen SP, Schilbach K, Rammensee HG, Salih HR, Jung G (2012) Generation, selection and preclinical characterization of an Fc-optimized FLT3 antibody for the treatment of myeloid leukemia. Leukemia 26(6):1228–1237
Hunger SP, Mullighan CG (2015) Acute lymphoblastic leukemia in children. N Engl J Med 373(16):1541–1552
Ju T, Cummings RD (2002) A unique molecular chaperone Cosmc required for activity of the mammalian core 1 beta 3-galactosyltransferase. Proc Natl Acad Sci U S A 99(26):16613–16618
Juszczynski P, Ouyang J, Monti S, Rodig SJ, Takeyama K, Abramson J, Chen W, Kutok JL, Rabinovich GA, Shipp MA (2007) The AP1-dependent secretion of galectin-1 by reed Sternberg cells fosters immune privilege in classical Hodgkin lymphoma. Proc Natl Acad Sci U S A 104(32):13134–13139
Kamemura K, Hayes BK, Comer FI, Hart GW (2002) Dynamic interplay between O-glycosylation and O-phosphorylation of nucleocytoplasmic proteins: alternative glycosylation/phosphorylation of THR-58, a known mutational hot spot of c-Myc in lymphomas, is regulated by mitogens. J Biol Chem 277(21):19229–19235
Kamper P, Ludvigsen M, Bendix K, Hamilton-Dutoit S, Rabinovich GA, Moller MB, Nyengaard JR, Honore B, d'Amore F (2011) Proteomic analysis identifies galectin-1 as a predictive biomarker for relapsed/refractory disease in classical Hodgkin lymphoma. Blood 117(24):6638–6649
Keiko K, Keigo A, Kei T, Nobuyuki T (2009) Loss of p53 enhances catalytic activity of IKKbeta through O-linked beta-N-acetyl glucosamine modification. Proc Natl Acad Sci U S A 106(9):3431–3436
Kirwan A, Utratna M, O’Dwyer ME, Joshi L, Kilcoyne M (2015) Glycosylation-based serum biomarkers for cancer diagnostics and prognostics. Biomed Res Int 2015:490531
Kniep B, Monner DA, Burrichter H, Diehl V, Muhlradt PF (1983) Gangliotriaosylceramide (asialo GM2), a glycosphingolipid marker for cell lines derived from patients with Hodgkin’s disease. J Immunol 131(3):1591–1594
Kobayashi T, Kuroda J, Ashihara E, Oomizu S, Terui Y, Taniyama A, Adachi S, Takagi T, Yamamoto M, Sasaki N, Horiike S, Hatake K, Yamauchi A, Hirashima M, Taniwaki M (2010) Galectin-9 exhibits anti-myeloma activity through JNK and p38 MAP kinase pathways. Leukemia 24(4):843–850
Kojima N, Hakomori S (1989) Specific interaction between gangliotriaosylceramide (Gg3) and sialosyllactosylceramide (GM3) as a basis for specific cellular recognition between lymphoma and melanoma cells. J Biol Chem 264(34):20159–20162
Komada Y, Sakurai M (1994) Shedding of CD9 antigen in acute lymphoblastic leukemia. Leuk Lymphoma 12(5–6):365–372
Koning MT, Quinten E, Zoutman WH, Kielbasa SM, Mei H, van Bergen CAM, Jansen P, Vergroesen RD, Willemze R, Vermeer MH, Tensen CP, Veelken H (2019) Acquired N-linked glycosylation motifs in B-cell receptors of primary cutaneous B-cell lymphoma and the normal B-cell repertoire. J Invest Dermatol 139(10):2195–2203
Kriss CL, Pinilla-Ibarz JA, Mailloux AW, Powers JJ, Tang CH, Kang CW, Zanesi N, Epling-Burnette PK, Sotomayor EM, Croce CM, Del Valle JR, Hu CC (2012) Overexpression of TCL1 activates the endoplasmic reticulum stress response: a novel mechanism of leukemic progression in mice. Blood 120(5):1027–1038
Krysov S, Potter KN, Mockridge CI, Coelho V, Wheatley I, Packham G, Stevenson FK (2010) Surface IgM of CLL cells displays unusual glycans indicative of engagement of antigen in vivo. Blood 115(21):4198–4205
Labdon JE, Gibson KD, Sun S, Pestka S (1984) Some species of human leukocyte interferon are glycosylated. Arch Biochem Biophys 232(1):422–426
Lafouresse F, Bellard E, Laurent C, Moussion C, Fournie JJ, Ysebaert L, Girard JP (2015) L-selectin controls trafficking of chronic lymphocytic leukemia cells in lymph node high endothelial venules in vivo. Blood 126(11):1336–1345
Larrue C, Saland E, Vergez F, Serhan N, Delabesse E, Mansat-De Mas V, Hospital MA, Tamburini J, Manenti S, Sarry JE, Recher C (2015) Antileukemic activity of 2-deoxy-d-glucose through inhibition of N-linked glycosylation in acute myeloid leukemia with FLT3-ITD or c-KIT mutations. Mol Cancer Ther 14(10):2364–2373
Li Y, Luo S, Dong W, Song X, Zhou H, Zhao L, Jia L (2016) Alpha-2, 3-sialyltransferases regulate the multidrug resistance of chronic myeloid leukemia through miR-4701-5p targeting ST3GAL1. Lab Invest 96(7):731–740
Linley A, Krysov S, Ponzoni M, Johnson PW, Packham G, Stevenson FK (2015) Lectin binding to surface Ig variable regions provides a universal persistent activating signal for follicular lymphoma cells. Blood 126(16):1902–1910
Ma W, Du J, Chu Q, Wang Y, Liu L, Song M, Wang W (2011) hCLP46 regulates U937 cell proliferation via Notch signaling pathway. Biochem Biophys Res Commun 408(1):84–88
Ma H, Cheng L, Hao K, Li Y, Song X, Zhou H, Jia L (2014a) Reversal effect of ST6GAL 1 on multidrug resistance in human leukemia by regulating the PI3K/Akt pathway and the expression of P-gp and MRP1. PLoS One 9(1):e85113
Ma L, Shan Y, Bai R, Xue L, Eide CA, Ou J, Zhu LJ, Hutchinson L, Cerny J, Khoury HJ, Sheng Z, Druker BJ, Li S, Green MR (2014b) A therapeutically targetable mechanism of BCR-ABL-independent imatinib resistance in chronic myeloid leukemia. Sci Transl Med 6(252):252ra121
Manzella L, Tirro E, Pennisi MS, Massimino M, Stella S, Romano C, Vitale SR, Vigneri P (2016) Roles of interferon regulatory factors in chronic myeloid leukemia. Curr Cancer Drug Targets 16(7):594–605
Marjon KD, Termini CM, Karlen KL, Saito-Reis C, Soria CE, Lidke KA, Gillette JM (2016) Tetraspanin CD82 regulates bone marrow homing of acute myeloid leukemia by modulating the molecular organization of N-cadherin. Oncogene 35(31):4132–4140
Marth JD, Grewal PK (2008) Mammalian glycosylation in immunity. Nat Rev Immunol 8(11):874–887
Massimino M, Consoli ML, Mesuraca M, Stagno F, Tirrò E, Stella S, Pennisi MS, Romano C, Buffa P, Bond HM, Morrone G, Sciacca L, Di Raimondo F, Manzella L, Vigneri P (2014) IRF5 is a target of BCR-ABL kinase activity and reduces cml cell 2 proliferation. Carcinogenesis 35(5):1132–1143
Maxson JE, Gotlib J, Pollyea DA, Fleischman AG, Agarwal A, Eide CA, Bottomly D, Wilmot B, McWeeney SK, Tognon CE, Pond JB, Collins RH, Goueli B, Oh ST, Deininger MW, Chang BH, Loriaux MM, Druker BJ, Tyner JW (2013) Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical CML. N Engl J Med 368(19):1781–1790
Maxson JE, Luty SB, MacManiman JD, Abel ML, Druker BJ, Tyner JW (2014) Ligand independence of the T618I mutation in the colony-stimulating factor 3 receptor (CSF3R) protein results from loss of O-linked glycosylation and increased receptor dimerization. J Biol Chem 289(9):5820–5827
Maxson JE, Luty SB, MacManiman JD, Paik JC, Gotlib J, Greenberg P, Bahamadi S, Savage SL, Abel ML, Eide CA, Loriaux MM, Stevens EA, Tyner JW (2016) The Colony-stimulating factor 3 receptor T640N mutation is oncogenic, sensitive to JAK inhibition, and mimics T618I. Clin Cancer Res 22(3):757–764
Mazurov D, Ilinskaya A, Heidecker G, Filatov A (2012) Role of O-glycosylation and expression of CD43 and CD45 on the surfaces of effector T cells in human T cell leukemia virus type 1 cell-to-cell infection. J Virol 86(5):2447–2458
Mondal S, Chandra S, Mandal C (2010) Elevated mRNA level of hST6Gal I and hST3Gal V positively correlates with the high risk of pediatric acute leukemia. Leuk Res 34(4):463–470
Moremen KW, Tiemeyer M, Nairn AV (2012) Vertebrate protein glycosylation: diversity, synthesis and function. Nat Rev Mol Cell Biol 13(7):448–462
Mugnaini EN, Ghosh N (2016) Lymphoma. Prim Care 43(4):661–675
Mullighan CG (2012) Molecular genetics of B-precursor acute lymphoblastic leukemia. J Clin Invest 122(10):3407–3415
Nakamura-Bencomo S, Gutierrez DA, Robles-Escajeda E, Iglesias-Figueroa B, Siqueiros-Cendon TS, Espinoza-Sanchez EA, Arevalo-Gallegos S, Aguilera RJ, Rascon-Cruz Q, Varela-Ramirez A (2021) Recombinant human lactoferrin carrying humanized glycosylation exhibits antileukemia selective cytotoxicity, microfilament disruption, cell cycle arrest, and apoptosis activities. Invest New Drugs 39(2):400–415
Nakayama R, Kuroda J, Taniyama N, Yamamoto-Sugitani M, Wada S, Kiyota M, Mizutani S, Chinen Y, Matsumoto Y, Nagoshi H, Shimura Y, Kobayashi T, Horiike S, Sato K, Taniwaki M (2014) Suppression of SERPINA1-albumin complex formation by galectin-3 overexpression leads to paracrine growth promotion of chronic myelogenous leukemia cells. Leuk Res 38(1):103–108
Nangalia J, Green AR (2017) Myeloproliferative neoplasms: from origins to outcomes. Blood 130(23):2475–2483
Nasirikenari MVL, Collins CC et al (2014) Remodeling of marrow hematopoietic stem and progenitor cells by non-self ST6Gal-1 Sialyltransferase. J Biol Chem 289(10):7178–7189
Niemann CU, Kjeldsen L, Ralfkiaer E, Jensen MK, Borregaard N (2007) Serglycin proteoglycan in hematologic malignancies: a marker of acute myeloid leukemia. Leukemia 21(12):2406–2410
Nowell PC (2002) Progress with chronic myelogenous leukemia: a personal perspective over four decades. Annu Rev Med 53:1–13
Odabashian M, Carlotti E, Araf S, Okosun J, Spada F, Gribben JG, Forconi F, Stevenson FK, Calaminici M, Krysov S (2020) IGHV sequencing reveals acquired N-glycosylation sites as a clonal and stable event during follicular lymphoma evolution. Blood 135(11):834–844
Ogretmen B, Hannun YA (2004) Biologically active sphingolipids in cancer pathogenesis and treatment. Nat Rev Cancer 4(8):604–616
Ohki K, Kiyokawa N, Saito Y, Hirabayashi S, Nakabayashi K, Ichikawa H, Momozawa Y, Okamura K, Yoshimi A, Ogata-Kawata H, Sakamoto H, Kato M, Fukushima K, Hasegawa D, Fukushima H, Imai M, Kajiwara R, Koike T, Komori I, Matsui A, Mori M, Moriwaki K, Noguchi Y, Park MJ, Ueda T, Yamamoto S, Matsuda K, Yoshida T, Matsumoto K, Hata K, Kubo M, Matsubara Y, Takahashi H, Fukushima T, Hayashi Y, Koh K, Manabe A, Ohara A, Tokyo Children’s Cancer Study Group (2019) Clinical and molecular characteristics of MEF2D fusion-positive B-cell precursor acute lymphoblastic leukemia in childhood, including a novel translocation resulting in MEF2D-HNRNPH1 gene fusion. Haematologica 104(1):128–137
Ouyang J, Plutschow A, Pogge von Strandmann E, Reiners KS, Ponader S, Rabinovich GA, Neuberg D, Engert A, Shipp MA (2013) Galectin-1 serum levels reflect tumor burden and adverse clinical features in classical Hodgkin lymphoma. Blood 121(17):3431–3433
Pang X, Li H, Guan F, Li X (2018) Multiple roles of glycans in hematological malignancies. Front Oncol 8:364
Patel AB, O’Hare T, Deininger MW (2017) Mechanisms of resistance to ABL kinase inhibition in chronic myeloid leukemia and the development of next generation ABL kinase inhibitors. Hematol Oncol Clin North Am 31(4):589–612
Patel K, Danilov AV, Pagel JM (2019) Duvelisib for CLL/SLL and follicular non-Hodgkin lymphoma. Blood 134(19):1573–1577
Peng L, Zhang Y, Xin H (2020) lncRNA SNHG3 facilitates acute myeloid leukemia cell growth via the regulation of miR-758-3p/SRGN axis. J Cell Biochem 121(2):1023–1031
Petrini M, Pelosi-Testa E, Sposi NM, Mastroberardino G, Camagna A, Bottero L, Mavilio F, Testa U, Peschle C (1989) Constitutive expression and abnormal glycosylation of transferrin receptor in acute T-cell leukemia. Cancer Res 49(24 Pt 1):6989–6996
Pinho SS, Reis CA (2015) Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer 15(9):540–555
Pirro M, Schoof E, van Vliet SJ, Rombouts Y, Stella A, de Ru A, Mohammed Y, Wuhrer M, van Veelen PA, Hensbergen PJ (2019) Glycoproteomic analysis of MGL-binding proteins on acute T-cell leukemia cells. J Proteome Res 18(3):1125–1132
Pluchart C, Barbe C, Poitevin G, Audonnet S, Nguyen P (2021) A pilot study of procoagulant platelet extracellular vesicles and P-selectin increase during induction treatment in acute lymphoblastic leukaemia paediatric patients: two new biomarkers of thrombogenic risk? J Thromb Thrombolysis 51(3):711–719. https://doi.org/10.1007/s11239-020-02346-7
Preyer M, Vigneri P, Wang JY (2011) Interplay between kinase domain autophosphorylation and F-actin binding domain in regulating imatinib sensitivity and nuclear import of BCR-ABL. PLoS One 6(2):e17020
Price A, Druhan LJ, Lance A, Clark G, Vestal CG, Zhang Q, Foureau D, Parsons J, Hamilton A, Steuerwald NM, Avalos BR (2020) T618I CSF3R mutations in chronic neutrophilic leukemia induce oncogenic signals through aberrant trafficking and constitutive phosphorylation of the O-glycosylated receptor form. Biochem Biophys Res Commun 523(1):208–213
Radcliffe CM, Arnold JN, Suter DM, Wormald MR, Harvey DJ, Royle L, Mimura Y, Kimura Y, Sim RB, Inoges S, Rodriguez-Calvillo M, Zabalegui N, de Cerio AL, Potter KN, Mockridge CI, Dwek RA, Bendandi M, Rudd PM, Stevenson FK (2007) Human follicular lymphoma cells contain oligomannose glycans in the antigen-binding site of the B-cell receptor. J Biol Chem 282(10):7405–7415
Ratajczak MZ, Adamiak M (2015) Membrane lipid rafts, master regulators of hematopoietic stem cell retention in bone marrow and their trafficking. Leukemia 29(7):1452–1457
Reilly JT (2003) FLT3 and its role in the pathogenesis of acute myeloid leukaemia. Leuk Lymphoma 44(1):1–7
Reis CA, Osorio H, Silva L, Gomes C, David L (2010) Alterations in glycosylation as biomarkers for cancer detection. J Clin Pathol 63(4):322–329
Ren R (2005) Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer 5(3):172–183
Rowley JD, Potter D (1976) Chromosomal banding patterns in acute nonlymphocytic leukemia. Blood 47(5):705–721
Ruvolo PP (2019) Galectins as regulators of cell survival in the leukemia niche. Adv Biol Regul 71:41–54
Ruvolo PP, Ruvolo VR, Benton CB, AlRawi A, Burks JK, Schober W, Rolke J, Tidmarsh G, Hail N Jr, Davis RE, Andreeff M (2016) Combination of galectin inhibitor GCS-100 and BH3 mimetics eliminates both p53 wild type and p53 null AML cells. Biochim Biophys Acta 1863(4):562–571
Ruvolo PP, Ruvolo VR, Burks JK, Qiu Y, Wang RY, Shpall EJ, Mirandola L, Hail N Jr, Zeng Z, McQueen T, Daver N, Post SM, Chiriva-Internati M, Kornblau SM, Andreeff M (2018) Role of MSC-derived galectin 3 in the AML microenvironment. Biochim Biophys Acta Mol Cell Res 1865(7):959–969
Sackstein R, Dimitroff CJ (2000) A hematopoietic cell L-selectin ligand that is distinct from PSGL-1 and displays N-glycan-dependent binding activity. Blood 96(8):2765–2774
Sadigh S, Hasserjian RP, Hobbs G (2020) Distinguishing atypical chronic myeloid leukemia from other Philadelphia-negative chronic myeloproliferative neoplasms. Curr Opin Hematol 27(2):122–127
Scarfo L, Ferreri AJ, Ghia P (2016) Chronic lymphocytic leukaemia. Crit Rev Oncol Hematol 104:169–182
Schmidt-Arras DE, Bohmer A, Markova B, Choudhary C, Serve H, Bohmer FD (2005) Tyrosine phosphorylation regulates maturation of receptor tyrosine kinases. Mol Cell Biol 25(9):3690–3703
Schoch C, Kern W, Kohlmann A, Hiddemann W, Schnittger S, Haferlach T (2005) Acute myeloid leukemia with a complex aberrant karyotype is a distinct biological entity characterized by genomic imbalances and a specific gene expression profile. Genes Chromosomes Cancer 43(3):227–238
Sedlik C, Heitzmann A, Viel S, Ait Sarkouh R, Batisse C, Schmidt F, De La Rochere P, Amzallag N, Osinaga E, Oppezzo P, Pritsch O, Sastre-Garau X, Hubert P, Amigorena S, Piaggio E (2016) Effective antitumor therapy based on a novel antibody-drug conjugate targeting the Tn carbohydrate antigen. Onco Targets Ther 5(7):e1171434
Shi Y, Tomic J, Wen F, Shaha S, Bahlo A, Harrison R, Dennis JW, Williams R, Gross BJ, Walker S, Zuccolo J, Deans JP, Hart GW, Spaner DE (2010) Aberrant O-GlcNAcylation characterizes chronic lymphocytic leukemia. Leukemia 24(9):1588–1598
Silva ML (2015) Cancer serum biomarkers based on aberrant post-translational modifications of glycoproteins: clinical value and discovery strategies. Biochim Biophys Acta 1856(2):165–177
Skliris A, Labropoulou VT, Papachristou DJ, Aletras A, Karamanos NK, Theocharis AD (2013) Cell-surface serglycin promotes adhesion of myeloma cells to collagen type I and affects the expression of matrix metalloproteinases. FEBS J 280(10):2342–2352
Spiciarich DR, Oh ST, Foley A, Hughes SB, Mauro MJ, Abdel-Wahab O, Press RD, Viner R, Thompson SL, Chen Q, Azadi P, Bertozzi CR, Maxson JE (2018) A novel germline variant in CSF3R reduces N-glycosylation and exerts potent oncogenic effects in leukemia. Cancer Res 78(24):6762–6770
Stagno F, Stella S, Spitaleri A, Pennisi MS, Di Raimondo F, Vigneri P (2016) Imatinib mesylate in chronic myeloid leukemia: frontline treatment and long-term outcomes. Expert Rev Anticancer Ther 16(3):273–278
Stella S, Tirro E, Conte E, Stagno F, Di Raimondo F, Manzella L, Vigneri P (2013) Suppression of survivin induced by a BCR-ABL/JAK2/STAT3 pathway sensitizes imatinib-resistant CML cells to different cytotoxic drugs. Mol Cancer Ther 12(6):1085–1098
Stowell SR, Ju T, Cummings RD (2015) Protein glycosylation in cancer. Annu Rev Pathol 10:473–510
Sugawara S, Im C, Kawano T, Tatsuta T, Koide Y, Yamamoto D, Ozeki Y, Nitta K, Hosono M (2017) Catfish rhamnose-binding lectin induces G0/1 cell cycle arrest in Burkitt’s lymphoma cells via membrane surface Gb3. Glycoconj J 34(1):127–138
Tanaka TN, Bejar R (2019) MDS overlap disorders and diagnostic boundaries. Blood 133(10):1086–1095
Taniguchi N, Miyoshi E, Ko JH, Ikeda Y, Ihara Y (1999) Implication of N-acetylglucosaminyltransferases III and V in cancer: gene regulation and signaling mechanism. Biochim Biophys Acta 1455(2–3):287–300
Tringali C, Lupo B, Cirillo F, Papini N, Anastasia L, Lamorte G, Colombi P, Bresciani R, Monti E, Tettamanti G, Venerando B (2009) Silencing of membrane-associated sialidase Neu3 diminishes apoptosis resistance and triggers megakaryocytic differentiation of chronic myeloid leukemic cells K562 through the increase of ganglioside GM3. Cell Death Differ 16(1):164–174
Tripodo C, Florena AM, Macor P, Di Bernardo A, Porcasi R, Guarnotta C, Ingrao S, Zerilli M, Secco E, Todaro M, Tedesco F, Franco V (2009) P-selectin glycoprotein ligand-1 as a potential target for humoral immunotherapy of multiple myeloma. Curr Cancer Drug Targets 9(5):617–625
Turzanski J, Grundy M, Shang S, Russell N, Pallis M (2005) P-glycoprotein is implicated in the inhibition of ceramide-induced apoptosis in TF-1 acute myeloid leukemia cells by modulation of the glucosylceramide synthase pathway. Exp Hematol 33(1):62–72
Vainer H, Bussel A (1976) Membrane glycoproteins and platelet protein glycosylation in chronic myeloid leukemia. Nouv Rev Fr Hematol 16(3):447–454
Vuillier F (2005) Lower levels of surface B-cell-receptor expression in chronic lymphocytic leukemia are associated with glycosylation and folding defects of the μ and CD79a chains. Blood 105(7):2933–2940
Wang Y, Chang N, Zhang T, Liu H, Ma W, Chu Q, Lai Q, Liu L, Wang W (2010) Overexpression of human CAP10-like protein 46 KD in T-acute lymphoblastic leukemia and acute myelogenous leukemia. Genet Test Mol Biomarkers 14(1):127–133
Wang S, Itoh M, Shiratori E, Ohtaka M, Tohda S (2018) NOTCH activation promotes glycosyltransferase expression in human myeloid leukemia cells. Hematol Rep 10(3):7576
Wiels J, Holmes EH, Cochran N, Tursz T, Hakomori S (1984) Enzymatic and organizational difference in expression of a Burkitt lymphoma-associated antigen (globotriaosylceramide) in Burkitt lymphoma and lymphoblastoid cell lines. J Biol Chem 259(23):14783–14787
Williams AB, Li L, Nguyen B, Brown P, Levis M, Small D (2012) Fluvastatin inhibits FLT3 glycosylation in human and murine cells and prolongs survival of mice with FLT3/ITD leukemia. Blood 120(15):3069–3079
Wu JL, Chiang MF, Hsu PH, Tsai DY, Hung KH, Wang YH, Angata T, Lin KI (2017) O-GlcNAcylation is required for B cell homeostasis and antibody responses. Nat Commun 8(1):1854
Yamamoto-Sugitani M, Kuroda J, Ashihara E, Nagoshi H, Kobayashi T, Matsumoto Y, Sasaki N, Shimura Y, Kiyota M, Nakayama R, Akaji K, Taki T, Uoshima N, Kobayashi Y, Horiike S, Maekawa T, Taniwaki M (2011) Galectin-3 (Gal-3) induced by leukemia microenvironment promotes drug resistance and bone marrow lodgment in chronic myelogenous leukemia. Proc Natl Acad Sci U S A 108(42):17468–17473
Yang WH, Kim JE, Nam HW, Ju JW, Kim HS, Kim YS, Cho JW (2006) Modification of p53 with O-linked N-acetylglucosamine regulates p53 activity and stability. Nat Cell Biol 8(10):1074–1083
Yao D, Huang Y, Huang X, Wang W, Yan Q, Wei L, Xin W, Gerson S, Stanley P, Lowe JB, Zhou L (2011) Protein O-fucosyltransferase 1 (Pofut1) regulates lymphoid and myeloid homeostasis through modulation of Notch receptor ligand interactions. Blood 117(21):5652–5662
Yelvington BJ (2018) Subcutaneous rituximab in follicular lymphoma, chronic lymphocytic leukemia, and diffuse large B-cell lymphoma. J Adv Pract Oncol 9(5):530–534
Yen SC, Chen LC, Huang HL, Ngo ST, Wu YW, Lin TE, Sung TY, Lien ST, Tseng HJ, Pan SL, Huang WJ, Hsu KC (2021) Investigation of selected flavonoid derivatives as potent FLT3 inhibitors for the potential treatment of acute myeloid leukemia. J Nat Prod 84(1):1–10
Yoon PS, Hyun Sil K, Nam Hee K, Suena J, So Young C, Jeong Gu K, Ichiro O, Keiji S, Noboru K, Hyung Wook N (2014) Snail1 is stabilized by O-GlcNAc modification in hyperglycaemic condition. EMBO J 29(22):3787–3796
Yoshimura M, Nishikawa A, Ihara Y, Nishiura T, Nakao H, Kanayama Y, Matuzawa Y, Taniguchi N (1995) High expression of UDP-N-acetylglucosamine: beta-D mannoside beta-1,4-N-acetylglucosaminyltransferase III (GnT-III) in chronic myelogenous leukemia in blast crisis. Int J Cancer 60(4):443–449
Yoshimura M, Ihara Y, Ohnishi A, Ijuhin N, Nishiura T, Kanakura Y, Matsuzawa Y, Taniguchi N (1996) Bisecting N-acetylglucosamine on K562 cells suppresses natural killer cytotoxicity and promotes spleen colonization. Cancer Res 56(2):412–418
Yu Y, Kou D, Liu B, Huang Y, Li S, Qi Y, Guo Y, Huang T, Qi X, Jia L (2020) LncRNA MEG3 contributes to drug resistance in acute myeloid leukemia by positively regulating ALG9 through sponging miR-155. Int J Lab Hematol 42(4):464–472
Zhang X, Dong W, Zhou H, Li H, Wang N, Miao X, Jia L (2015) Alpha-2,8-sialyltransferase is involved in the development of multidrug resistance via PI3K/Akt pathway in human chronic myeloid leukemia. IUBMB Life 67(2):77–87
Zhou H, Li Y, Liu B, Shan Y, Li Y, Zhao L, Su Z, Jia L (2017) Downregulation of miR-224 and let-7i contribute to cell survival and chemoresistance in chronic myeloid leukemia cells by regulating ST3GAL IV expression. Gene 626:106–118
Zhu D, McCarthy H, Ottensmeier CH, Johnson P, Hamblin TJ, Stevenson FK (2002) Acquisition of potential N-glycosylation sites in the immunoglobulin variable region by somatic mutation is a distinctive feature of follicular lymphoma. Blood 99(7):2562–2568
Zinzani PL, Rambaldi A, Gaidano G, Girmenia C, Marchetti M, Pane F, Tura S, Barosi G (2019) Infection control in patients treated for chronic lymphocytic leukemia with ibrutinib or idelalisib: recommendations from Italian society of hematology. Leuk Res 81:88–94
Acknowledgement
The authors thank Dr. Huadong Liu for critical reading and English editing of the manuscript. This work was supported by the National Natural Science Foundation of China (31971053, 81770123), China Postdoctoral Science Foundation, the Scientific and Technical Foundation of Shaanxi Province (2020JM015) and the Fundamental Research Funds for the Central Universities from Xi’an Jiaotong University.
Declaration of Competing Interest: The authors declare no competing financial interests.
Author Contributions: Y. C. and X. L. conceived and designed the article frame. H. S., M. W., X. P., F. G., X. L., and Y. C. wrote the paper.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Su, H., Wang, M., Pang, X., Guan, F., Li, X., Cheng, Y. (2021). When Glycosylation Meets Blood Cells: A Glance of the Aberrant Glycosylation in Hematological Malignancies. In: Pedersen, S.H.F. (eds) Reviews of Physiology, Biochemistry and Pharmacology. Reviews of Physiology, Biochemistry and Pharmacology, vol 180. Springer, Cham. https://doi.org/10.1007/112_2021_60
Download citation
DOI: https://doi.org/10.1007/112_2021_60
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-83429-6
Online ISBN: 978-3-030-83430-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)