
Abstract
There are an increasing number of recombinant antibodies and proteins in preclinical and clinical development for therapeutic applications. Mammalian expression systems are key to enabling the production of these molecules, and Chinese hamster ovary (CHO) cell platforms continue to be central to delivery of the stable cell lines required for large-scale production. Increasing pressure on timelines and efficiency, further innovation of molecular formats and the shift to new production systems are driving developments of these CHO cell line platforms. The availability of genome and transcriptome data coupled with advancing gene editing tools are increasing the ability to design and engineer CHO cell lines to meet these challenges. This chapter aims to give an overview of the developments in CHO expression systems and some of the associated technologies over the past few years.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Cell engineering
- Cell line development
- Chinese hamster ovary cells
- Gene editing
- Therapeutic protein production
1 Introduction
1.1 Why Mammalian Cells?
Mammalian cells are used for the production of recombinant monoclonal antibodies (mAbs) and complex proteins because they have the capacity to assemble and fold complex polypeptides and to perform post-translational modifications (PTMs) which are important for therapeutic bioactivity and bioavailability. Production processes using mammalian cell cultures in bioreactors are high yielding (up to 10 g/L for mAbs) and scalable (up to tens of thousands of litres), making them compatible with large-scale manufacture for clinical supply of therapeutic proteins. Transgenic animal systems can also produce complex proteins and offer some advantages in terms of cost and scale of supply over mammalian cell culture systems [1]. However, the timelines to establish transgenic herds or colonies are significantly longer than those for establishing cell culture systems and there are concerns regarding the theoretical transmission of xenotropic viruses to humans.
Mammalian cell culture expression systems rely on the introduction of vector DNA encoding the recombinant protein into a host cell line and harnessing the synthetic capacity of the cell to express and secrete the encoded protein into the cell culture medium. Systems for large-scale production of therapeutic proteins are generally based on stable recombinant cell lines created by integration of linearized plasmid DNA encoding the therapeutic protein into the host genome, so that the transgenes are transmitted to each daughter cell at cell division. Traditionally, the production process from a stable cell line is performed using the controlled culture conditions in a bioreactor using a fed-batch mode, with additional nutrients being “fed” into the bioreactor to sustain cell growth and productivity for the duration of the culture period. The recombinant protein is then recovered from the cell culture medium.
2 Choice of Mammalian Host Cell Lines
Mammalian host cell lines are able to perform PTMs including glycosylation, carboxylation, hydroxylation, sulfation and amidation, which can be important for biological activity [2]. A number of different mammalian host cell lines are used for large-scale production of complex therapeutic proteins (reviewed by Butler and Spearman [3]). Historically, these have been based on rodent cell lines – mouse myeloma (NS0 and Sp2/0), baby hamster kidney and CHO cells. Although these cell hosts are able to produce glycoproteins with human-like glycosylation profiles, they also produce non-human glycoform structures which can impact in vivo clearance and immunogenicity [4, 5].
A number of human host cell lines can be used for the production of recombinant proteins with fully human PTMs, as reviewed by Dumont et al. [6] and Swiech et al. [7]. Cell lines generated from the human embryonic kidney cell line HEK-293 are used for the production of approved therapeutic proteins, such as recombinant clotting factors and fusion proteins, where additional PTMs such as gamma-carboxylation and sulfation are required for bioactivity. The human fibrosarcoma cell line HT-1080 is used for the manufacture of approved enzyme therapies – iduronate-2-sulfatase, agalsidase alfa and velaglucerase alfa. The PER.C6 cell line, derived from human embryonic retinal cells [8], and the CAP-T cell line, derived from human amniocytes [9], have been employed to produce therapeutic proteins currently in preclinical and clinical development. Engineered human leukemic cell lines have been developed for the production of therapeutic proteins with fully human and optimised glycosylation [10, 11]. In addition, a human neuronal cell line, AGE1.HN, is being used for production of proteins with complex glycosylation profiles [12].
3 CHO Host Cells
Over several decades, the CHO host cell line has become established as an industry-standard expression platform with a strong regulatory track record, and it accounts for the production of >70% of approved therapeutic proteins [13]. Owing to the rodent origin of CHO cells, there is a species barrier to the production of viruses able to infect humans, and studies have confirmed that CHO cells are resistant to infection by many viruses that can infect humans [14]. Significant advances have been made in the productivity of CHO bioreactor processes through upstream process development, particularly with respect to the development of media and feed formulations [15]. This optimisation has resulted in robust and scalable bioreactor processes, achieving high cell densities and product yields, with titres of the order of 10 g/L for mAbs being attainable in fed-batch culture at scales of tens of thousands of litres. Importantly, not only can CHO cells be engineered with genes encoding therapeutic proteins but also further cell and genetic engineering can be used to modify cell line characteristics, such as growth and metabolism, as well as product quality attributes (reviewed by Fischer et al. [16]). Therefore, CHO cells provide a flexible expression platform that can be engineered to fit both process and product requirements. This engineering approach for CHO cells has been facilitated by the availability of genome sequences for CHO host cell lines [17, 18] and the recent advances in genome editing [16]. In light of the central importance of CHO cell systems to the biopharmaceutical industry, the remainder of this review focuses on the recent developments to CHO production systems.
3.1 Development of CHO Expression Systems
There are a variety of drivers for further developing CHO expression platforms for therapeutic protein production:
-
Efficiency and timelines. Discovery platforms are becoming more efficient in identifying multiple leads with different modes of action, and at the same time there is pressure to advance projects rapidly into the clinic. As the creation of the stable manufacturing cell line is a pre-requisite for the production of clinical material, it is desirable to reduce timelines for cell line development and even parallel track cell line development for multiple molecules to enable project acceleration to critical-path GLP toxicology and to the clinic (reviewed by Estes and Melville [19]).
-
Innovation of novel therapeutic proteins. Following the success of engineered antibodies, proteins and fusion proteins as therapeutics, biological activities are now being combined to create novel bispecific molecules. These non-natural molecules can pose challenges for development because of their often low expression yield, need for more-complex PTMs and other product quality attributes such as aggregation. Although it is preferable that these undesirable characteristics are screened out during the discovery process, this is not always possible. Therefore, engineering of the production cell line and/or process is needed to improve the ability to manufacture these molecules.
-
Manufacturing processes. The pressure to reduce cost of goods and to maximise the efficiency of production capacity and facilities is driving manufacturing processes towards new process paradigms such as continuous processing [20, 21]. Continuous upstream processes involve higher cell densities in the bioreactor and longer culture times, creating unique demands on the performance parameters of the production cell line, such as cell metabolism and production stability, compared with those for traditional fed-batch processes.
3.2 CHO Cell Line Diversity and Evolution
There are a number of different CHO host cell lines, as reviewed by Wurm [22] and Lewis [17]. The first CHO cell line was derived from the ovary of an adult Chinese hamster [23] and later underwent cloning and other manipulations to generate different cell lines, including CHO-K1, CHO DG44, CHO-S and CHO DUXB11. These CHO cell lines were originally cultured in media containing animal serum, but, because of concerns about the cost of serum, batch-to-batch variation in serum performance in culture media and the potential for adventitious agent contamination, these cell lines have been adapted to grow in culture media that are free from serum or any other animal-derived components. The choice of a CHO host cell line is partly driven by the compatibility with the expression plasmid selection system used for recombinant protein production. The CHO DG44 cell line is deficient in dihydrofolate reductase (DHFR) and so is typically used with the DHFR selectable marker that can complement this deficiency. The other commonly used CHO expression system is based on using glutamine synthetase (GS) as the selectable marker and is generally used with host cell lines derived from CHO-K1.
Different CHO host cell lines can exhibit differences in productivity. Hu and co-workers demonstrated that recombinant cell lines from a CHOK1 host showed higher productivities for two difficult-to-express (DTE) mAbs compared with cell lines constructed using a DUXB11 host [24]. Similarly, auditioning of DG44 and CHOK1 cell lines with an artificial chromosome carrying copies of genes for a recombinant mAb showed differences in performance, with cell lines derived from the CHOK1 host showing higher productivity [25]. However, it is difficult to make direct comparisons between different hosts as the performance of the cell line is also strongly affected by the process conditions, including media and feed composition, which can be optimised to improve individual cell line performance. In addition, the CHO host cell lines are themselves heterogeneous, containing a population of cells that show variation in growth, metabolism, biosynthetic capacity and ability to perform PTMs [26, 27].
The phenotypic variation of CHO cells results from the underlying genetic and epigenetic diversity. The genetic heterogeneity can be observed at a gross level as the varied karyology profiles of individual cells in a host population with different chromosomal structures [22, 28]. This chromosomal variation arises from dynamic genome restructuring which occurs during continuous subculture and is characteristic of immortalized cell lines. It is the combination of genomic and epigenetic remodelling at cell division that contributes to the versatility of CHO as a host cell line with the ability to adapt to different culture media and conditions, and to generate recombinant cell lines that express proteins with varying product quality profiles.
The phenotypic and genotypic variation within CHO cell populations can be exploited to isolate host cells with more desirable characteristics by serially sub-culturing cells in the presence of physical or chemical stresses that can select for desired properties. A striking example of this “directed evolution” approach is the use of plant cytotoxic lectins that recognise specific glycoform structures to select for host cells with modified glycosylation pathways – the Lec mutants (reviewed by Patnaik and Stanley [29]). On binding to specific glycoproteins at the cell surface, the lectins are internalized, whereupon they can exert their toxic effects, resulting in cell death. Cells that do not display the reciprocal glycoform structures, because of mutations caused naturally or by treatment with mutagenic agents, are able to survive the lectin treatment. The use of lectins with different specificities has allowed the identification of cell lines with different glycosylation mutations, which in turn have contributed to the elucidation of glycosylation pathways and associated genes as well as glycosylation engineering [29]. An example where a desirable bioprocessing characteristic was selected is described by Bort and colleagues [30], in which CHO cells were sequentially cultured in medium containing stepwise-reduced levels of glutamine. The cells able to survive each reduction in glutamine were recovered by fluorescence-activated cell sorting (FACS), and the final population of selected cells was able to grow in glutamine-free medium. This follows on from the work of Prentice and co-workers [31] where DG44 host cells were selected for their ability to survive in bioreactor conditions, leading to increases in peak cell density and the ability to grow in the absence of growth factors. Similarly, bioreactor evolution and selection may provide a strategy to generate host cell lines that are more suited for continuous upstream processes. To be able to take advantage of an evolved phenotype in the host cell line, it must be maintained over the timescales needed for cell line development and manufacture. This can require continued application of the selective pressure used to derive the phenotype or the screening of individual cell lines for stability of the desired characteristic without selection.
4 Vector Engineering
Currently, conventional non-viral expression plasmids containing transgenes are still the major vector platform for cell line development. These plasmids contain multiple expression cassettes, each consisting of a promoter and associated regulatory elements to drive transcription, the coding sequences of the recombinant protein and selectable marker, and a sequence for transcript termination and polyadenylation. The recombinant protein gene encodes a homologous or heterologous N-terminal secretory leader peptide to direct the protein for secretion via the endoplasmic reticulum (ER) and the Golgi, where PTMs, such as glycosylation, take place. Following transfection of the plasmid DNA into the host cell line, stable transfectants are generated through the application of drug to select for the expression of the selectable marker gene. Standard plasmid transfection processes result in random integration of the vector into the host genome, and the site of integration along with copy number of the vector influence the level of transgene expression. Therefore, extensive transfectant screening needs to be performed to identify high-expressing cell lines. Expression vectors have been optimised to increase the productivity and stability of cell lines and to improve the efficiency of the cell line generation process. These vector optimisation approaches have included manipulation of selection markers, promoter engineering, incorporation of new DNA regulatory elements, the usage of different codons to regulate translation, and modulation of the order and ratio of expression of different gene cassettes, some of which are described in more detail below. Meanwhile, novel vector platforms for targeted integration and transposon-based vector systems have been developed to increase integration efficiency.
4.1 Manipulation of Selection Markers
There are two main selection systems used to generate production CHO cell lines, and these are based on the metabolic genes encoding DHFR and GS that are typically selected by the respective addition of the inhibitors methotrexate (MTX) and methionine sulfoximine (MSX) to the cell culture medium [32, 33]. As the selectable marker and the gene(s) encoding the recombinant protein are usually combined on the same vector, integration of the vector into a genome location favourable for selectable marker transcription is also generally beneficial for the expression of the linked recombinant protein genes. Therefore, a high stringency of selection facilitates obtaining cell lines possessing a high level of transcription from integration of the expression vector at an active locus in the genome and also for removing any low producers.
The CHO-DG44 and DUKXB11 hosts are DHFR deficient and require addition of glycine, hypoxanthine and thymidine (GHT) to the culture medium for cell growth. Integration and expression of the DHFR gene complements the DHFR deficiency of the CHO host cell line, allowing growth in the absence of GHT. Furthermore, higher levels of DHFR expression and the linked transgenes can be selected by stepwise increases in the levels of MTX, which is a highly selective competitive inhibitor of DHFR. Gene amplification resulting from chromosomal remodelling is a naturally occurring phenomenon in CHO cells, and the increased level of MTX selects for cells that have undergone amplification of the copies of the DHFR marker gene loci, which can also include the recombinant protein genes. However, this amplification process is laborious and time-consuming, and the multiple tandem vector repeats that result from the amplification process can be unstable, leading to a loss of productivity over time [34]. An alternative approach to increasing gene copy number to enhance transgene transcription is to attenuate the expression level of the DHFR selectable marker. Thus, only cells from the most transcriptionally active loci survive the selection. Marker attenuation can be achieved in a number of ways. It has been reported that codon de-optimisation that decreased the translation efficiency of the DHFR gene resulted in approximately threefold higher production of an Fc fusion protein [35]. The addition of the AU-rich elements in the 3′ untranslated region (3′ UTR) of the DHFR gene to reduce mRNA half-life and/or the inclusion of the murine ornithine decarboxylase (MODC) PEST amino acid sequence to promote DHFR protein degradation were shown to improve the specific productivities for recombinant human interferon gamma in DG44 cells [36]. Another approach to de-optimising DHFR expression by placing the DHFR gene downstream of an attenuated internal ribosome entry (IRES) element allowed the production of high levels of the small soluble glycoprotein Dectin-1 [37]. By combining the engineered PEST motif with an attenuated IRES sequence, the DHFR protein level was further reduced and resulted in increased recombinant alpha-1 antitrypsin production [38].
The GS gene encodes glutamine synthetase, which catalyses the conversion of glutamate to glutamine. As glutamine is an essential amino acid, GS expression is required for cells to grow in glutamine-free medium. However, CHO cells naturally express GS in glutamine-free medium, so the use of GS as a selectable marker requires the addition of the competitive inhibitor MSX to the cell culture medium. The addition of MSX ensures that only those cells producing higher levels of GS resulting from expression of the GS selectable marker on the plasmid vector can survive in the selective conditions [32]. Efforts to increase the selection strength of the GS gene have mostly focussed on cell line engineering and optimisation of the transfection and selection processes. It has been shown that knocking out the endogenous GS genes in the CHO host with zinc finger nuclease (ZFN) technology resulted in multiple cells lines with higher sensitivity to MSX selection and yielded a sixfold increase in the frequency of high producers for a recombinant antibody, thereby providing the potential to improve the efficiency of the cell line development screening process [39]. Suppression of endogenous GS gene expression by increasing the glutamine concentration in the cell culture medium before transfection has also proved to be an efficient way to increase the strength of selection with the same concentration of MSX [40].
4.2 Multi-Gene Expression with Novel Promoters and Elements
Production of mAbs requires the co-expression of the heavy- and light-chain genes along with the selection marker. Often, all three genes are incorporated into a single vector to ensure co-expression of the physically linked genes. However, the development of novel multi-unit bispecific antibodies, as well as large enzyme complexes and DTE proteins that require co-expression of genes encoding specific chaperones and PTM enzymes, necessitates the co-expression of multiple transgenes. Incorporating multi-gene expression cassettes into a single plasmid is technically challenging because of size restrictions of standard plasmids, both in terms of plasmid construction and propagation in Escherichia coli as well as the efficient transfection and integration of larger plasmids into the CHO host cell line. In addition, repeated use of the same promoter for the expression of multiple genes on a single plasmid can cause promoter interference [41], which may limit expression.
One approach to avoid repeated promoter sequences is to utilize different promoters for each gene cassette. In addition to the commonly used human cytomegalovirus immediate early (hCMVIE) promoter, there is a range of viral and housekeeping promoters, such as those derived from the simian virus 40 (SV40) and the human elongation factor 1 alpha gene (EF1α), which can be used for protein production. Further CHO endogenous promoters with desirable expression profiles have been identified by utilizing transcriptomics data [42]. To expand the search beyond natural promoters, a synthetic-biology approach has been applied to construct a library of synthetic promoters by combining different transcription factor regulatory elements (TFREs) from powerful viral promoters [43]. Screening of the synthetic promoters from this library by evaluation in transient transfections has identified promoters with transcriptional activity ranging over two orders of magnitude, some significantly exceeding that of the hCMVIE promoter. The use of a strong synthetic promoter has the potential to improve gene expression and the use of multiple promoters of varying strength could more precisely control the relative expression of different genes encoding multi-subunit proteins, which might be advantageous for protein expression and product quality [44,45,46,47]. Synthetic promoters are also shorter than conventional promoters, reducing the size of vectors with multi-gene cassettes and thereby improving vector handling and transfection efficiency.
Another approach for removing repeated promoters is to drive transcription of linked multiple genes as a single transcript from a single promoter. The insertion of an IRES element between each coding region, or cistron, allows ribosomes to initiate translation at multiple points along the transcript and so different polypeptides can be translated from the same transcript. As the translation of the gene downstream of an IRES sequence is through a weaker CAP-independent mechanism, it usually results in a lower level of expression of the second and any subsequent cistron. This can create an imbalance in the production of two linked subunits which might not be desirable for some molecules [48]. However, for other molecules, changing the proportion of the different subunits can be beneficial [44,45,46,47]. In the 2A technology, multiple linked genes are translated as a single open reading frame. The coding sequences of the different genes are separated by motifs encoding the self-cleaving viral 2A peptide sequence. This enables the production of equimolar ratios of component subunits from the single precursor polypeptide. The 2A self-processing peptide system has been used for antibody production and has shown a twofold increase in transient expression compared with the equivalent IRES-linked construct for the same antibody [44, 45]. Viral 2A peptides from different viruses, such as foot-and-mouth disease virus, equine rhinitis A virus, porcine teschovirus-1 and Thosea asigna virus, have been used for mAb production [49]. None of these 2A peptides produced complete cleavage, but adding a glycine–serine linker provided more flexibility at the boundary between two linked chains and thus enhanced the cleavage [49]. The insertion of a furin recognition site upstream of the 2A peptide sequence allowed additional sequence-specific protein cleavage and the removal of 2A residues that otherwise remained attached to the upstream heavy chain protein [49].
5 Vector Integration
Despite efforts to optimise plasmid-based vector systems, reliable and efficient integration of transgenes into transcriptionally active genomic loci still remains a major challenge for stable protein expression. As productive integration events are rare, extensive cell line screening is required to identify and characterise the desired high producers. In recent years, the frequency and/or efficiency of productive integration events has been increased by including chromosomal elements on the expression plasmid vector or by combining with transposon or targeted integration technologies. Viral-based integration systems such as lenti- and baculovirus-mediated gene delivery technologies are also being developed for the efficient expression of secreted proteases and membrane glycoproteins [50, 51].
5.1 Incorporation of Chromosomal Elements
A number of chromosomal elements that have a positive effect on promoting high-level and stable gene expression have been incorporated into plasmid vectors. These include nuclear scaffold/matrix attachment regions (S/MARs) [52] and ubiquitous chromatin opening elements (UCOEs) [53]. These chromosomal architectural elements affect the adjacent chromatin structure once the plasmid vector has been integrated into the genome to maintain accessibility of the vector DNA for transcription and prevent gene silencing. Recent work has suggested that CHO cell lines generated with UCOE-containing vectors not only showed resistance to chromosomal position effects with increased mRNA production per copy of transgene but also grew to a higher cell density [54]. The UCOE system is versatile having been used in combination with multiple selection and amplification platforms, in different CHO host cell lines and with the high-throughput Clonepix screening process [54,55,56]. Its beneficial effect on the frequency of higher-expressing cell lines and robustness of cell growth allows the rapid generation of stable transfectant pools and has been used to replace transient transfection for the rapid production of cytokines and other recombinant proteins [57]. Saunders and co-workers [58] compared a number of chromatin structural elements including UCOE, MAR, STAR (Stabilising Anti Repressor) and cHS4 (an insulator from the chicken beta-globin locus control region) for their ability to confer resistance to insertional position effects that could decrease mAb expression. UCOE had the most beneficial effect of all the elements tested, maintaining a high level of expression and showing reduced promoter methylation, which is one cause of gene silencing.
5.2 Transposon-Based Vector Systems
Transposons are a class of naturally occurring non-viral mobile genetic elements that have the ability to integrate single copies of DNA sequences with high frequency at multiple loci within the host genome [59]. Transposon DNA vectors have been developed for a variety of purposes, including insertional mutagenesis as well as gene transfer and therapy. Typically, these transposon systems have two components: a donor plasmid with the cargo transgenes flanked by the transposon inverted repeat sequences and a helper plasmid or mRNA encoding a transposase. The transposase is transiently expressed from the helper plasmid or mRNA and then catalyses the excision of the inverted repeat sequence flanked region of the donor plasmid and facilitates its integration into the host genome. Transposon vectors have been deployed with CHO cell lines for the production of recombinant proteins including a gamma-secretase integral membrane protease complex [60]. The Piggyback (PB) transposon, a class II transposable element originally derived from the cabbage looper moth, has been favoured in the bio-production field because of ease of handling and its capability for mobilizing very large DNA sequences, such as bacterial artificial chromosomes [61]. However, the frequency of transposition decreases as the size of the artificial transposon increases beyond 14 kb. In a side-by-side comparison, Matasci and colleagues [62] showed that the PB transposon system resulted in 15–20 times more recombinant cells in the transfectant population and that the derived clonal cell lines had higher average volumetric productivity and greater production stability than cell lines originating from the standard plasmid vector. Based on this result, the group utilized the PB transposon system for rapid transfectant pool generation to produce high titres of an antibody and a human tumour necrosis factor receptor-Fc (TNFR-Fc) fusion protein [63]. The PB transposon pools expressing TNFR-Fc fusion protein had a constant volumetric productivity for up to 3 months in the absence of selection. Further optimisation of the PB transposon system has been performed by incorporating a human MAR sequence into the PB vector, which significantly increased transgene integration and transcription [64]. This study also showed that, with the PB transposon system, transfectants can readily be generated without selection, and high levels of expression could be obtained from as few as 2–4 genomic copies of the MAR-containing transposon vector. The attributes of low transgene copy number and stability in the absence of selection that are conferred by the PB transposon system are highly desirable for production cell lines as they are associated with transgene stability over long-term culture. Moreover, the higher productivity and the increased frequency of productive cell lines are highly beneficial for the efficiency of the cell line development process.
5.3 Targeted Integration
Integration at a predefined chromosomal locus that gives homogeneous, high expression is advantageous for protein production and the efficiency of cell line generation. In addition, because cells with the same isogenic background are expected to have similar and predictable growth and metabolism, this approach is also beneficial for upstream process development [65]. Site-specific recombinase systems such as Cre-Lox and Flp-FRT have been the common tools used for targeted integration (reviewed by Bode et al. [66] and Turan et al. [67]). These systems are usually operated in two steps: first, following random integration, screening and tagging expression hotspots using a reporter gene vector that also contains a recombination-specific sequence tag (Lox or FRT) and then, second, targeting integration of a vector containing the cargo gene(s) and complementary recombination-sequences to the pre-tagged locus using transient expression of the recombination enzyme. Several groups have successfully demonstrated the generation of homogeneous cell lines expressing recombinant protein with good productivity and long-term production stability by using this approach [68, 69]. However, this is a lengthy process, and, as only a single copy of the transgenes is integrated, the resulting cell lines tend to have lower titres than the best cell lines derived from a random integration approach. In addition, as fluorescent reporter genes are not secreted, the tagged cell lines that are selected are not necessarily proficient in the production of secreted proteins.
To speed up the hotspot screening step and integrate an increased number of transgene copies, two technologies, ϕC31 integrase and CRE-Lox recombinase systems, have been combined [70]. The ϕC31 integrase mediates attB-specific DNA integration into the CHO genome at pseudo-attP sites. As there is a limited number (100–1,000) of pseudo-attP sites in the CHO genome, the scale of the first step of searching and tagging (with LoxP integration sites) of transcriptionally active pseudo attP spots is manageable. Moreover, it has been shown that targeted integration of two copies of antibody genes doubled the titres compared with targeted integration of one copy of the genes. Meanwhile, Zhang and colleagues [71] have used a vector containing mAb genes flanked with recombination sequences for large-scale screening to identify cell lines with good productivity, long-term production stability and possession of low-copy number transgenes. After removing the mAb genes through recombinase-mediated cassette exchange (RMCE) using an Flp-FRT-containing null cassette, the resulting host was used for the efficient and consistent construction of cell lines possessing high mAb productivity (2–2.5 g/L in fed-batch shake flasks) and stability of expression for more than 100 generations. This approach not only identified transcriptional hotspots for integration but also generated host cells with intrinsic production capability and stability that was inherited from the progenitor cell lines.
In addition to naturally occurring site-specific enzymes, such as Cre, Flp and ϕC31, which are capable of recognising specific sequences and then promoting the interchange between two recombination sites, a number of programmable sequence-specific nucleases that generate double-stranded breaks (DSBs) have been applied to genome editing in mammalian cells. The first of these programmable reagents were the ZFNs where protein engineering is used to enable targeting of double-strand DNA cleavage adjacent to chosen DNA sequences (reviewed by Chandrasegaran and Carroll [72]). Typically, the chromosomal DSBs introduced by ZFNs are repaired by a non-homologous end joining (NHEJ) repair pathway, in which the DSBs are ligated without the use of a homologous template. In some instances, the DNA joining repair is imprecise and leads to a deletion or insertion of nucleotides, causing a frameshift that can result in gene disruption. By designing and engineering a nuclease target site into the donor plasmid vector, Cristea and colleagues [73] showed that a ZFN can cleave both donor and chromosome DNA to produce efficient integration of the donor plasmid into the CHO genome through a non-homologous end joining (NHEJ) pathway.
The transcription activator-like effector nucleases (TALENs) are another class of programmable site-specific nucleases. TALENs consist of two domains, an engineered TALE that binds to a specific DNA sequence and a DNA cleavage domain. It has been demonstrated that a large expression cassette that includes a gene encoding a single-chain Fv-Fc (scFv-Fc) can be knocked in at a predefined locus in the CHO genome mediated by a TALEN with micro-homology to the targeted locus [74, 75]. The simplicity of the vector construction that requires no large regions of homology along with the efficiency of the process for isolating knock-in cell lines is advantageous for the generation of production cell lines.
More recently, RNA-guided nucleases, based on the CRISPR–Cas9 system from prokaryotes, have been developed and are being widely used for genome editing, including for CHO cells (reviewed by Lee et al. [76, 77]). Cas9 is an endonuclease that uses a guide RNA to target specifically cleavage of DNA sequences that are complementary to the guide RNA. Unlike ZFNs and TALENs, which require complex protein engineering to cleave new DNA target sequences, the CRISPR–Cas9 system uses a universal DNA endonuclease and cleavage specificity is engineered by simply modifying the sequence of the guide RNAs. Therefore, the CRISPR–Cas9 system significantly increases the efficiency and reduces the cost of the design and generation of the reagents for genome engineering. Furthermore, engineering of a CRISPR–Cas9 recognition site into a donor plasmid can promote NHEJ-based integration of transgenes into a predefined locus, albeit at a low efficiency [78]. The efficiency of NHEJ targeted integration remains low even with the aid of promoter trapping (HEK293) or phenotypic screening (HPRT- in CHO) strategies at 0.17% and 0.45%, respectively. However, using a transgene cassette flanked by homology arms in the presence of locus-specific guide RNAs and Cas9 protein enables more efficient integration (10.2–27.8%) into multiple pre-defined loci in the CHO genome through a homology-directed repair mechanism [76, 77]. The application of the CRISPR–Cas9 system for targeted integration shows benefits in terms of the consistency of transgene expression in the resulting cell lines. In addition, the insert capacity for multiple gene cassettes (~5 kb) and the increasing targeting efficiency mediated by the CRISPR–Cas9 system advocate its development as a targeted integration platform for production cell line generation. However, challenges remain as off-target effects, presumably because of non-targeted integration, have the potential to affect cellular functions in the engineered cells.
Targeted integration technologies provide advantages for the rapidity of cell line development and also the potential homogeneity and consistency of cell line productivity. These inherent benefits are exploited by the deployment of targeted integration as a research tool and for rapid supply of early preclinical and clinical supply of therapeutic proteins. However, a key drawback of targeted integration systems for commercial manufacturing is the lower productivity compared with that of cell lines derived from conventional random integration and screening approaches. The lower expression results from the lower transgene copy number and also the lack of epigenetic selection for high expression for targeted integration compared with random integration. These factors are being addressed by ongoing developments to enable targeted integration of multiple transgene cassettes and also by “reusing” a high-yielding, stable, random-integration production cell line by removing and replacing the product genes with a suitable targeting cassette, as demonstrated with an RMCE system by Zhang et al. [71]. However, there is a concern as to whether a single clonal targeted integration host can possess all the intrinsic properties, genetic and epigenetic, to generate the desired product quality characteristics for all molecular formats. Therefore, a toolbox of host clones might be required, with different host clones for different products.
6 Glycoengineering
Glycosylation is the enzymatic addition of carbohydrate (glycans) and is one of the most important PTMs for therapeutic proteins as it can affect biological activity, stability, pharmacokinetics and immunogenicity. N-linked and O-linked glycosylation are the most common types of protein glycosylation, with the pathways for N-linked glycosylation being the best characterised in mammalian cells (reviewed by Hossler et al. [79]). The sites for glycosylation are determined by the structure of the protein, and the protein conformation can also affect glycan structures. The host cell line and the cell culture conditions also influence the glycan structures and the glycan homogeneity [79]. This can result in heterogeneous glycan profiles and, because of the potential impact on biological activity, there can be a need to demonstrate consistent lot-to-lot glycosylation depending on the mode of action of the molecule [80]. Although CHO cell lines generally produce human-like glycans, they can also produce some non-human glycoform structures (NGNA and non-human alpha-gal) which are undesirable from an immunogenicity perspective. The natural N-glycosylation profiles from CHO cells can be improved by cell line engineering to produce more-homogeneous and more-desirable glycosylation profiles (reviewed by Dicker and Strasser [81]). A “glycodelete engineering” strategy to produce simplified and homogeneous glycan structures has been developed in HEK 293 cells [82]. This approach could be beneficial for production of mAbs from CHO in which the mode of action is antigen neutralization without effector function. Recent work on engineering the N-linked glycoforms in the CHO cells most relevant for therapeutic recombinant proteins is described below.
6.1 Terminal Sialylation
Sialylation plays important roles in the half-life of therapeutic proteins. The hepatic asialoglycoprotein receptor (ASPR) can recognise terminal galactose residues and mediates serum protein degradation. Terminal sialylation can hide the galactose from recognition; thus sialylated proteins are cleared more slowly than those that are asialylated. Consequently, it is highly desirable to control and increase the level of sialylation, but recombinant proteins from CHO cells tend not to be fully sialylated. Therefore, sialylation pathways are genetic engineering targets for improving the extent and/or consistency of recombinant protein sialylation profiles.
There are different ways to attach sialic acid to galactose: human proteins predominantly use the α2,6-linkage, whereas CHO cells have incomplete α2,3-linked sialic acid. This difference is because of the lack of significant expression of the α2,6-sialyltransferase gene in CHO cell lines [17]. Early work showed the feasibility of using genetic engineering approaches to enhance sialylation of recombinant glycoproteins secreted from CHO cell lines by co-expressing α2,6-sialyltransferase or α2,3-sialyltransferase in recombinant CHO cell lines [83, 84]. More recently, a CHO-K1 host cell line has been engineered to express the hamster ST6GAL1 gene, which encodes α2,6-sialyltransferase [85]. Antibody produced in the engineered host not only showed the human-like α2,6-linked terminal sialic acid, but also a twofold increase in the overall sialylation level compared with that of the unmodified host. Similarly, recent work conducted by Yin and co-workers [86] showed that overexpression of the human ST6GAL1 gene in CHO cell lines producing human erythropoietin (EPO) resulted in increased sialylation. Furthermore, co-expression of two additional glycosyltransferases, α1,3-d-mannoside β1,4-N-acetylglucosaminyltransferase (GnTIV/Mgat4) and UDP-N-acetylglucosamine: α1,6-d-mannoside β1,6-N-acetylglucosaminyl transferase (GnTV/Mgat5) in the ST6GAL1-modified CHO cells, produced further enhancement of the terminal branching. As a result, tri- and tetra-antennary N-glycans represented approximately 92% of the total N-glycans on the resulting EPO protein. RNAi knock-down experiments have been conducted to investigate further which of the six CHO α2,3-sialyltransferases (ST3GAL1-ST3GAL6) with their different substrate specificities are critical for alpha 2,3-sialyation linkage of CHO glycoproteins [87]. Results indicated that ST3GAL3, ST3GAL4 and ST3GAL6 are involved in N-linked sialylation and ST3GAL4 may play a vital role in glycoprotein sialylation of complex glycoproteins such as EPO. This study demonstrated the power of RNAi as a screening tool to identify individual and combinatorial effects of multiple genes in the glycosylation pathway and to provide targets for successful glycoengineering.
6.2 High-Mannose Glycans
High-mannose glycans are known to increase antibody immunogenicity and decrease half-life, making them undesirable for therapeutic proteins [88]. Nevertheless, proteins with mannose-only glycans are advantageous for X-ray crystallography studies because of the simple and homogeneous glycan structure [89, 90]. The MGAT1 gene product, also called GnTI, catalyses the transfer of N-acetylglucosamine to the Man5GlcNAc2 (Man5) N-glycan structure as part of complex N-glycan synthesis. Disruption of the MGAT1 gene either by chemical mutagenesis followed by lectin selection [91] or ZFN-mediated targeted knock-out (KO) technology [92] in multiple CHO cell lines resulted in the production of protein with Man5 as the predominant N-linked glycosylation species. Unlike the chemical mutagenesis method, ZFN mediates precise genomic modifications, so that the growth and productivity of the resulting KO cell lines are not adversely affected by random mutagenesis throughout the genome. The MGAT1 KO host is also useful in the production of mannose-terminated enzymes, such as recombinant glucocerebrosidase to treat patients with Gaucher disease, as the terminal mannose residues bind with better efficiency to the mannose receptor on the surface of the target macrophage cells [93]. Interestingly, by re-introducing the MGAT1 gene into the mgat1 mutant or KO cell lines, two independent groups have shown that the sialylation levels of IgG1 molecules were improved as well [85, 94,95,96]. This phenomenon was not observed when the MGAT1 transgene was expressed in wild-type CHO K1 cells. Although the exact mechanism is not well understood, this strategy of restoring the MGAT1 function in deficient cells has been applied in CHO cells from transient expression through to stable and large-scale perfusion systems to produce EPO with a greater proportion of tri- and tetra-antennary sialylation [94, 95].
6.3 Afucosylation for Increased Antibody-Dependent Cell-Mediated Cytotoxicity
Fucosylation remains a major target for glycoengineering as afucosylated mAbs have enhanced ADCC activities and an increased anti-tumour activity. Knocking out the FUT8 transferase gene through traditional homology-based recombination approaches [97] or ZFN-mediated gene disruption [98] has been shown to produce completely afucosylated antibodies. The recent development of the CRISPR–Cas9 technology has significantly increased the efficiency of gene editing, and it has been reported that the triple gene targets FUT8, BAK and BAX can be knocked out in a one-step manipulation to produce a FUT8-deficient host with anti-apoptotic properties [99]. This work demonstrated the multiplexing capability of the CRISPR–Cas9 system for genome editing with high efficiency.
Besides the FUT8 gene, many other genes in the fucosylation pathway have become engineering targets. Haryadi and colleagues [100] used a ZFN to inactivate the GDP-fucose transporter gene (Slc35c1) in a cell line with an existing mutation in the CMP-sialic-acid transporter gene (Slc35a1). This resulted in a cell line (CHO-gmt5) that produced afucosylated and asialylated mAbs. These investigators also compared ZFN, TALEN and CRISPR–Cas9 technologies for the modification of the slc35c1 gene locus and found changes in mAb titre in cell lines engineered with ZFN and CRISPR–Cas9, but not with TALEN, suggesting that TALEN might have fewer off-target effects.
A novel approach to engineering the fucosylation pathway is that of “biosynthetic deflection”. Von Horsten and colleagues [101] described the expression of a bacterial enzyme, GDP-4-keto-6-deoxymannose, to divert an intermediate substrate from the fucose synthesis pathway. This resulted in the production of afucosylated mAbs even when the expression levels of the bacterial gene were relatively low.
6.4 O-Glycoengineering
In contrast to N-linked glycosylation, the capabilities of CHO cells for O-linked glycosylation of proteins are less well understood. However, work using a “SimpleCell” strategy has been used to increase knowledge of O-glycoproteins and sites of O-glycan attachments in the CHO proteome [102]. This approach used a ZFN to knock out a component of the O-glycan pathway, leading to homogenous and truncated O-glycans and allowing enrichment of O-linked glycan proteins for identification by mass spectrometry. Data analysis from the study indicates that CHO cells have a limited capacity for O-glycosylation, which supports transcriptome studies also showing expression of a limited number of O-glycosylation GalNAc transferases [18]. Consequently, cell engineering approaches have the potential to improve O-linked glycosylation, which is important for PTMs in molecules such EPO and Etanercept (TNF alpha receptor-Fc-fusion).
7 New Formats and “Difficult-to-Express” Proteins
More recently, new classes of proteins such as multi-specific antibody and fusion proteins have been designed as therapeutic proteins for unmet medical needs. These novel formats can pose more challenges to mammalian cell expression systems compared with conventional mAbs. They can be poorly expressed and show undesirable levels of aggregation because of a combination of the intrinsic properties of these proteins and the limited biosynthetic capacity of the host cell lines for these heterologous proteins. In addition, there are some mAbs and “natural” molecules that also fall into this class of DTE proteins. Although high levels of transcription are required for high levels of protein expression, steps downstream from transcription are also important in regulating protein secretion from mammalian cells. These post-transcriptional steps include mRNA translation, translocation of polypeptides from the cytosol into the ER, polypeptide folding and assembly, addition of PTMs and secretion. Depending on the individual recombinant protein, limitations in these steps can result in aggregation and low productivity of the desired product. Investigations have been focussed on understanding the bottlenecks that underlie the poor levels of expression of these proteins and addressing these bottlenecks with cell line and vector engineering tools. A summary of some of the successful approaches is given below and in Table 1.
7.1 Protein Trafficking, Assembly and Secretion
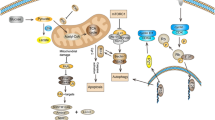
Secretory proteins have an N-terminal secretory peptide that targets the polypeptide for processing through the secretory machinery of the cell. As the nascent polypeptide emerges from the ribosome, the signal peptide binds to the signal recognition particle (SRP) and the resulting complex is targeted to the translocon on the ER membrane. As the polypeptide is translocated into the ER, the signal peptide is cleaved off by the signal peptidase so that the signal peptide is not part of the mature protein. Newly synthesized proteins are folded and assembled in the ER, before addition of PTMs and progression through the Golgi and final secretion. If the ER capacity for protein folding is exceeded, then the resulting unfolded or misfolded proteins accumulate in the ER, and this is detected and induces the unfolded protein response (UPR). The UPR aims to maintain protein homeostasis by shutting down translation or increasing the level of chaperones to aid folding (reviewed by Chakrabarti et al. [111]). At the same time, misfolded proteins are removed by upregulation of ER degradative enzymes. If ER stress is sustained, this can result in apoptosis and cell death. Where the production of unfolded or misfolded recombinant protein is contributing to significant ER stress then this naturally selects for low levels of productivity of the recombinant protein. The UPR is a dynamic and complex process, and two groups have developed UPR-responsive reporter systems in order to monitor and understand better the factors that can contribute to UPR stress [112, 113]. As highlighted below and in Table 1, components of the UPR, including chaperones, present potential engineering targets to improve cell line productivity for DTE proteins, as do strategies to reduce the levels of unfolded or misfolded protein.
Using an empirical modelling system to compare the transient expression of a panel of eight IgG1 molecules with a fourfold variation in volumetric productivity, Pybus and colleagues [105] determined that the mAb-specific expression limitation can be at the folding and assembly step. The DTE mAbs showed an induction of UPR in host CHO and a decrease in cell growth. By changing the ratio of heavy-chain (HC) and light-chain (LC) expression, and by co-expression of a variety of molecular chaperones, foldases or UPR transactivators (Table 1), the expression level of the DTE mAbs was significantly improved. A similar strategy and screening platform was used by Johari and colleagues [106] to investigate successfully the low productivity of an Fc-fusion protein (Sp35Fc), which was associated with the formation of intracellular oligomeric aggregates. By screening a panel of cellular and chemical chaperones and UPR transactivators, specific productivity and cell growth were manipulated and the productivity was increased by combinatorial approaches with reduced culture temperature (Table 1). An inducible system to express the spliced form of human X-box binding protein (XBPs) in combination with reduced temperature has also been used to increase mAb productivity [107]. Besides the manipulation of chaperones and the UPR pathway, the work from Le Fourn and co-workers [104] identified light-chain signal-peptide processing as the limiting step for the expression of a DTE antibody. The low level of mAb secretion was associated with an intracellular accumulation of unprocessed light chain that had retained the signal peptide. Overexpressing a human signal receptor protein, SRP14, and other components of the secretory pathway improved both the processing of the LC signal peptide and levels of mAb secretion. Other studies have shown that changing the secretory leader sequence can improve expression, although the mechanism underlying this effect is not well understood [114].
7.2 Aggregation
Protein aggregates are a concern for recombinant therapeutic proteins as they can impact efficacy as well as induce immunogenic responses and cause adverse events upon administration to patients. Therefore, there is a desire to minimize and control protein aggregation. By testing a tenfold range of ratios of LC to HC in stable CHO pools, Ho and colleagues [44, 45] have found that a higher ratio (>1) of LC:HC resulted in higher mAb titres and higher levels of monomer (Table 1). They also found that high-mannose-type N-glycans increased, whereas fucosylated and galactosylated glycans decreased significantly at the lowest LC:HC ratio. Further work by this group [46, 47] showed that the antibody aggregates consisted mostly of HC polypeptide, and, if cell pools producing higher levels of aggregate were re-transfected with the LC gene, then the level of aggregate was reduced. Overexpression of the BiP chaperone also reduced the level of aggregate, although the effect was less dramatic. It was found that the level of aggregation of an Fc-fusion protein was proportional to the gene dose in a HEK293 transient system [103]. As shown in Table 1, reducing input vector DNA and lowering the temperature significantly reduced mAb aggregation. It also increased the cleavage efficiency of a signal peptide, presumably because the reduced transcription rate allowed more time for cells to translate and process polypeptide. In a separate study of 28 individual mAb-expressing cell lines, the level of aggregate had an inverse correlation with intracellular and secreted light chain [115]. Another study of a bispecific antibody suggested a relationship between N-glycans and aggregation, with aggregate present in the cell culture medium containing antibody with reduced levels of N-glycan fucose and galactose residues [116]. Culture process development, such as optimisation of osmolarity and temperature, can also be used to reduce protein aggregation [117, 118]. Together these studies suggest that, depending on an individual recombinant protein, optimisation of the protein expression rate, the balance of expression of different subunits and the extent of glycosylation might be beneficial in reducing aggregation. High-throughput expression systems, such as that described by Hansen and colleagues [119], would potentially be useful in evaluating and optimising factors that influence these processes for an individual recombinant protein to improve expression and reduce aggregation.
7.3 Product-Related Cell Toxicity
There are several examples of recombinant proteins showing toxic effects in CHO cells, including reduced cell growth. Not surprisingly these proteins are difficult to express as the selection pressure generated by such toxicity leads to low productivity or productivity loss during cell expansion. Depending on the nature of the underlying interaction of these recombinant proteins with the CHO cell line, different mitigations have been identified to allow improved expression of these “toxic” products.
Misaghi et al. [108] observed up to a fourfold decrease in the expression of a mAb by clonal cell lines over 45 days of expansion, and the reduction was not associated with a decrease in heavy- and light-chain gene transcription. An inducible expression system was established that reduced the exposure of cells to the product, and, as a result, the stability of mAb productivity was maintained. In another example, cell line engineering proved to be a powerful tool in down-regulating a specific receptor to avoid the toxicity produced by expression of a bioactive ligand product. Romand and co-workers demonstrated that expression of human IGF-1 variants resulted in both poor growth and low productivity in CHO cells [109]. The negative effect of the IGF-1 product on cell growth was found to be mediated through the CHO IGF-1 receptor (IGF-1R). Consequently, by knocking out or knocking down the IGF-1R receptor gene in the CHO host cell lines, the productivity of recombinant IGF-1 in CHO cell lines was increased up to tenfold. In a cell line and process development case-study for another recombinant protein that bound to the cell surface and inhibited cell growth, one approach included selecting a host cell population that was able to grow in the presence of the recombinant protein in the culture medium [110]. Transfectant pools generated from the adapted host produced levels of the recombinant protein that were approximately threefold higher than those from the non-adapted host. Other approaches included using multiple host cell lines with diverse genetic composition, significantly increasing the bioreactor screen size during cell line development by using micro-bioreactors and also developing a modified cell culture medium to improve cell growth in the presence of the recombinant protein. This integrated approach resulted in a tenfold titre improvement.
8 Operating Existing Systems in New Ways
With the increasing emphasis on reducing timelines from lead discovery to clinical studies, there is a desire to accelerate the preclinical development phases, including toxicology studies. The availability of product of the appropriate quality is a limiting step for preclinical studies. To produce representative material, it is desirable to use the final cell line clone in the manufacturing bioreactor process. Progression to this stage of clonal cell line and process development can be expedited by the use of standard production platforms and processes, but it is still time consuming – in the order of at least 8 months from transfection to final clone and process. However, material can be made much earlier using transients or pools of cells, as outlined below. In considering this approach it is important to recognise the risks and impact if the product quality from the final clone and process is significantly different from that of the preclinical material. However, this risk can be mitigated by screening final cell lines and processes for product quality that matches that of the early material used for preclinical studies.
8.1 Transients
In contrast to stable expression, transient gene expression (TGE) does not require integration of the expression plasmid into the host cell genome and so no selection pressure is applied. The expression plasmid DNA is transfected into host cells; the DNA that reaches the nucleus is transcribed and the resulting transcript translated into protein, which is then secreted into the cell culture medium. The cells express the recombinant proteins encoded by the plasmid over a period of a few days to a few weeks. TGE has traditionally been used for the rapid production of recombinant proteins for use during discovery as research reagents as well as for the production of candidate molecules for evaluation and early characterisation. Although the capacity for high TGE yields from HEK 293 cells has been well established for many years [120], expression in CHO cells was limited until relatively recently. The product quality of recombinant proteins obtained from HEK and CHO cells can be different because of the differences in PTMs [121,122,123,124]. The desire to generate early material that is representative of later-stage processes that use stable CHO cell lines has driven the development of improved CHO transient expression systems. The strategies for enhancing transient expression have involved engineering the CHO host cell line, and developing the transient transfection and production processes with either wild-type or engineered CHO host cell lines. The various approaches for CHO transient expression development that have been documented in the literature have recently been reviewed by Jager and colleagues [125] and are summarised in Table 2. However, as a key driver for CHO transient system development is producing representative material of stable cell lines, industrialists have tended to set up optimised systems that are bespoke to their individual CHO host cell lines and production platforms, and consequently this information is not necessarily in the public domain. With the advent of the “Expi CHO” system there is now a commercially available CHO transient expression kit that is widely accessible (Table 2).
8.2 Transient Scale-Up
The strategies to improve CHO transient expression systems have culminated in the ability to express mAbs at 2 g/L in 21 days from transfection to harvest at a 6-L culture volume in a wave bioreactor using a process that is amenable to further scale-up [128]. Such high productivities at scale mean that it is now feasible to produce sufficiently large yields of recombinant proteins by transient expression to enable preclinical development and potentially even clinical development. However, sourcing the large amounts of plasmid DNA of the appropriate quality required for transient expression at large-scale is challenging and represents a different potential bottleneck for recombinant protein production compared with stable cell line systems. The inherent variability of transient transfections can also make the reproducibility of transient batches technically challenging.
8.3 Expression Predictability
Although it is now feasible to perform CHO transient expression at scale, the typical development process is to use a clonal cell line for the manufacture of clinical products. It typically takes several weeks to obtain readouts on expression from the stable cell line development process, which can cause project delays if there are issues with expression. However, the recent performance improvements in the CHO transient system allow prediction of the productivities in stable CHO cell lines as there is a correlation between CHO expression of mAbs in transient and stable expression formats. Therefore, CHO transient expression is also a valuable tool in screening different molecules during the discovery process to predict expression titres rapidly as well as producing batches of representative product for developability studies.
8.4 Stable Pools for Rapid Large-Scale Supply
In contrast to transient transfections at scale, the process for generating clonal stable cell lines requires much less plasmid DNA, but is both time and resource intensive as individual clones are recovered from single cells and then expanded for screening. An intermediate solution for more rapid production is to use transfectant pools of cells. Instead of screening individual transfectants, multiple transfectants are selected and recovered together as a pool, allowing a more rapid recovery of cell populations and therefore a more rapid scale-up for production. However, a transfectant pool is heterogeneous, containing a mixed population of cells with different levels of productivity that gives lower overall productivity than can be obtained from the best individual clonal cell lines. Furthermore, transfectant pools can show instability of expression over time. There are some technologies that promote more homogeneous transfectant expression, improving expression levels and expression stability; these include incorporation of UCOE sequences into expression vectors along with targeted integration and transposon-based systems, which were described in Sect. 5. These pool approaches are also compatible with the use of platform bioreactor processes that are typically used for clonal cell lines, which helps to ensure that the pool-derived product is representative of product from later-stage clonal cell lines.
8.5 Development of Cloning Technologies
Although feasible to make high yields of clinical grade recombinant proteins rapidly through scaled-up transients and transfectant pools, clonal production cell lines are central for commercial supply strategies because of their higher productivity and robustness for scale-up. Critical to the cell line development process is the regulatory guidance to isolate production cell lines from single progenitor cells [132] to ensure consistency of product quality. A number of different strategies and technologies are used to isolate clonal cell lines, with more recent developments focussing on reducing timelines and improving efficiency for cell line development. In limiting-dilution cloning, dilute cell suspensions are dispensed into multi-well plates at less than one cell per well and then cell lines are recovered from the single colonies that grow in individual wells. A statistical analysis of data on the recovery of colonies in wells and multiple rounds of cloning are used to support the clonality of the derived cell lines. More recently, the development of high-content plate imaging systems has allowed the generation of detailed images of the originating single cell in a well at the time of plating and can reduce the number of rounds of cloning required to support clonality. Another approach uses the ClonePix technology, which involves introducing low concentrations of cells into a semi-solid medium, allowing single cells to grow into colonies and then using the automated imaging and picking capabilities of the robot to transfer single, well-separated colonies into the individual wells of a multi-well plate [133]. Typically, ClonePix methods use two rounds of cloning to derive cell lines with a suitable assurance of clonality. Fluorescent detection reagents can be added to the semi-solid medium to allow identification of the colonies that are secreting recombinant product. FACS is an efficient technique for sorting cell suspensions and depositing single cells into individual wells of a multi-well plate. In combination with multi-well plate imaging of the deposited cells, FACS-based cloning requires only a single round of cloning [134] and is therefore more rapid for cell line development than methods requiring two rounds of cloning. In addition, the sorting capability of the FACS instrument can be harnessed by using fluorescently labelled detection reagents to bind either the product or a surrogate present on the cell surface, and then sorting on the basis of the bound fluorescence signal. New microfluidics technologies are also being applied to single-cell cloning [135]. Cell suspensions can be emulsified in oil, creating picodroplets that can then be imaged on microfluidic chips, with those containing a single cell being sorted and subsequently dispensed into plates. In addition, microfluidics provides the prospect of being able to couple isolation of single cells with performing assays on the picodroplet for secreted product to assess yield or product quality [136]. Single-cell cloning technologies based on “cell printing”, which involve microfluidic dispensing integrated with cell imaging and analysis, also show potential for cell line development applications [137, 138].
8.6 Improved Cell Line Screening
Stable transfection generates cell lines that show variation in productivity, growth and product quality. This diversity of characteristics arises from a combination of the random integration of the expression vector into the host genome, variation in transgene copy number and also from the phenotypic variation in individual cells in the host cell population, as discussed in Sect. 3.2 of this chapter [22, 27]. With the resulting recombinant cell line heterogeneity, it is important to incorporate the appropriate screens during the cell line development process to ensure the selection of candidate production cell lines with the appropriate growth, productivity and product quality attributes. Many of the recent advances in cell line screening are oriented towards increasing the efficiency of the cell line development process, often through automation, and enhancing the predictability of the cell line screening data of the performance of cell lines in bioreactors.
Key to the cell line development screening strategy is that cell lines are tested in a process representative of the platform bioreactor process using production medium and feed. Hence, cell lines are selected to “fit-to-process”, and this reduces the need for upstream process development before scale-up and clinical manufacture. During cell line development, multiple cell lines are evaluated to find those with suitable characteristics. To handle the large numbers of cell lines involved, this evaluation process involves a screening cascade with a series of cell line assessment steps where the numbers of cell lines reduce at each stage and at the same time the amount of characterisation data for each cell line increases. The first step identifies those cell lines expressing the recombinant protein usually by detecting product secreted into the culture medium. Expressing cell lines are then advanced to the next step that involves evaluating cell lines in fed-batch culture to assess growth and productivity. This was traditionally performed using shake flask cultures, but the laborious manual handling involved limits the number of cell lines that can be evaluated in parallel to a few tens. The development of high-throughput, small-scale, fed-batch culture processes using 24- or 96-well plates enables hundreds of cell lines to be assessed in parallel [139], with automation further reducing the manual handling effort required. Once the numbers of cell lines have been reduced to the top 24–48, scaled-down bioreactor systems that control pH and dissolved oxygen can be used to generate data that are predictive of larger-scale fed-batch bioreactors in terms of cell growth, productivity and metabolism [140, 141]. These microscale bioreactor systems are also being adapted to operate in a simulated perfusion mode, enabling the screening and identification of cell lines that are compatible with continuous upstream processes. Importantly, microscale bioreactors provide not only predictive bioreactor performance data but also product for the generation of representative product quality data; together these data are analysed to identify candidate production cell lines for further process characterisation, including cell line stability, before selecting the final clone for the creation of the master cell bank that is used for manufacture.
8.7 Product Characterisation During Cell Line Screening
Both the cell line and the upstream process used for therapeutic protein expression influence product quality attributes such as aggregation, fragmentation and PTMs. Product quality screening therefore needs to be incorporated into the cell line development process to ensure selection of cell lines that express product with suitable characteristics. The product quality attributes that are characterised are determined by the properties of the product itself, but typically include an assessment of glycosylation, aggregation, fragmentation and amino acid sequence integrity. The generation of analytical data for product from multiple cell lines during cell line development is facilitated by high-throughput analysis of product within the cell culture medium, for example for aggregation [142], or by integration with high-throughput purification methods, for example glycosylation assays [143]. Mass spectrometry and peptide mapping methods [144] are used to confirm that the product has the expected amino acid sequence. Product sequence variants that contain one or more amino acid substitutions can result from mutations in the encoding DNA or misincorporation of amino acids during translation [144,145,146,147]. As these sequence variants are cell line specific, they can be screened out during clone selection. Sequencing of cDNA can be used to characterise and confirm that the correct transcript sequence is expressed. However, this might not be sufficiently sensitive to identify low levels of a sequence variant, whereas next-generation sequencing (NGS) is more sensitive and can also provide additional data on transcript integrity [148,149,150].
8.8 Next-Generation Sequencing for Cell Line Characterisation
The level of annotation of the CHO and Chinese hamster genomes along with bioinformatics tools relating to the analysis of CHO omics data are continuing to be developed [151]. This facilitates the use of NGS to characterise the genomes of CHO cell lines. NGS allows detailed analysis of the genome following transgene integration or gene editing and can be used to assess transgene sequence, copy number, integrity and integration site. NGS is a very powerful technology, producing vast amounts of sequence data and it is essential to have the appropriate bioinformatics capabilities to process and analyse these data. Multiple targeted massive parallel sequencing (MPS) approaches have also been developed to focus on particular genomic regions defined by primers to reduce the scale of the data [152]. Other applications for NGS are to compare the transgene structure and integration at different times over long-term culture to assess the genetic stability of stable cell lines. Next-generation nucleic acid sequencing technology also provides an additional potential method for testing and investigating incidences of contamination [153]. As next-generation sequencing technology gains regulatory acceptance, it also has the potential to reduce the need for the in vivo testing that forms part of the traditional program of virus testing. Collaborative efforts involving regulators and cross-industry representatives are under way to investigate the sensitivity, robustness and validation of NGS methodologies for safety testing and to establish a framework for implementation [154, 155].
9 Perspectives on CHO Expression System Development
Traditionally, CHO cell line development for recombinant protein production has been a screening-led process using a sequential cascade of assays to identify cell lines with suitable characteristics in terms of growth, productivity and product quality for large-scale production. These screening processes mine the variation in cell line performance that arises from a combination of heterogeneity of cells in the host cell population and the heterogeneity resulting from random integration of the plasmid expression vector into the host cell genome. Many of the developments for CHO cell line generation have focussed on improving the predictability and efficiency of the screening processes, as described above. These systematic approaches have been very successful, leading to significant improvements in the timelines and the resources needed for cell line development, and in concert with intensive media development and bioreactor optimisation have delivered cell lines with higher productivities, achieving up to 10 g/L for mAbs. Although highly effective, these approaches have treated CHO cells as a “blackbox” with limited molecular understanding of the integrated networks of CHO biosynthetic processes. However, new challenges with increased requirements for efficiency in cell line development, expression of innovative molecular formats and new production processes require additional, more-rational design-led strategies to achieve the required optimisation and develop the next generation of CHO cell line development platforms.
The first publication of the genome sequence of CHO-K1 in 2011 [18] marked a shift in the level of molecular understanding in CHO cells and has stimulated more-rational engineering approaches to develop CHO production cell lines. Expression profiling of CHO cells and their responses to bioprocessing conditions have enabled a greater understanding of cellular processes and identified engineering targets to make improvements [156]. Furthermore, CHO cells can now be evaluated with multiple omics tools to describe the proteome and metabolome as well as the genome and the transcriptome, allowing the application of systems biology modelling [157]. Data from these omics approaches can be integrated with metabolic networks into computational “genome scale” metabolic models for specific pathways (reviewed by Gutierrez and Lewis [158]). These models are potentially very powerful, generating the ability to perform in silico experiments to predict the outcomes of changing components within biochemical pathways. Reported applications for these models to CHO cells have included media development and understanding the impact of different culture conditions [158]. Further integration of omics data to refine and build more-extensive models to incorporate biosynthetic processes is computationally challenging, but will enable more detailed and accurate predictions and further help to define engineering targets. Meanwhile, the collection and mapping of data from the CHO glycoproteome and phosphoproteome are improving the understanding of PTMs, which are important for the product quality of therapeutic proteins.
Having identified potential gene engineering targets, tools such as shRNA for gene knock-down and genome editing for gene knock-out along with standard plasmid vectors for gene knock-in or overexpression have been essential in validating and exploiting these gene targets. High-throughput screening expression systems such as that described by Hansen et al. [119] are potentially useful tools to explore rapidly the impact of combinations of genes on the expression of recombinant proteins and on product quality to validate potential targets. Programmable sequence-specific nucleases based on ZFN and TALEN technologies have shown great utility for knocking out single genes and in a few cases a combination of a small number of genes. However, the advent of the CRISPR–Cas9 system with its simpler and more rapid target engineering capacity means that multi-gene genome editing has become more feasible. Additional targeted genome editing applications for CRISPR–Cas9 include gene insertion and gene activation or repression as well as gene knock-out [76, 77], which are also important tools for creating optimal production lines.
Standard expression plasmid vectors and gene editing technologies offer a way to modulate individual or small numbers of genes involved in CHO cellular pathways. However, engineering of miRNAs can allow simultaneous modification of multiple genes across multiple pathways. MicroRNAs are small non-coding RNAs that are involved in regulating many cellular processes by a mechanism based on anti-sense recognition of specific sequences in target RNAs. The knowledge of the function of miRNAs is still developing, but they have been characterised as being involved in cell growth, apoptosis and cell death, hypoxia and oxidative stress as well as protein production (reviewed in Jadhav et al. [159]). Therefore, miRNAs are promising engineering targets to improve CHO production cell lines, and this is borne out by studies where overexpression of miR-17 and miR-30 produced higher expression levels of recombinant proteins from CHO cells [160, 161], although the molecular basis for these effects is not yet understood.
CHO cells can be conventionally engineered by accessing the natural diversity of genetic sequences from CHO cells or other organisms. However, synthetic biology approaches can devise and develop novel combinations of biological components and have the potential to redesign and improve CHO cellular processes radically for recombinant protein production (reviewed by Lienert et al. [162] and Xie and Fussenegger [163]). Through precise control of new gene networks, the resulting “designer cells” have the potential to improve production efficiency and robustness (e.g. through engineering metabolic and biosynthetic pathways), improve product homogeneity (e.g. through engineering pathways for PTMs) and enable the production of innovative and new molecular formats that are currently challenging to express. The building blocks for synthetic CHO-based systems are under development with the availability of libraries of new synthetic promoters to regulate transcription in CHO cells [164] and the development of new multi-gene engineering vectors to introduce new multi-gene synthetic networks into mammalian cells [165].
With the advantages of regulatory provenance, increasing knowledge of, and ability to manipulate, biosynthetic pathways and compatibility with new continuous manufacturing processes, CHO expression systems are set to become an even more flexible platform and are expected to continue to be central for delivery of increasingly complex therapeutic proteins. In future, it is envisioned that the data from omics technologies and integration with systems biology approaches will “open” the CHO “blackbox” and should enable a step change in the understanding and modelling of cellular processes in CHO cells to identify new rational engineering targets to improve recombinant protein production [166]. At the same time, genome editing in combination with synthetic biology technologies will provide the translational tools required to re-engineer CHO cells and exploit these targets. Together, these approaches should allow implementation of a more rational engineering and design-led approach to develop the next generation of CHO production cell lines tailored according to the product and process requirements.
References
Wang Y, Zhao S, Bai L, et al (2013) Expression systems and species used for transgenic animal bioreactors. Biomed Res Int 2013:580463. doi:10.1155/2013/580463
Walsh G, Jefferis R (2006) Post-translational modifications in the context of therapeutic proteins. Nat Biotechnol 24(10):1241–1252. doi:10.1038/nbt1252
Butler M, Spearman M (2014) The choice of mammalian cell host and possibilities for glycosylation engineering. Curr Opin Biotechnol 30:107–112. doi:10.1016/j.copbio.2014.06.010
Ghaderi D, Taylor RE, Padler-Karavani V, et al (2010) Implications of the presence of N-glycolylneuraminic acid in recombinant therapeutic glycoproteins. Nat Biotechnol 28(8):863–867. doi:10.1038/nbt.1651
Ghaderi D, Zhang M, Hurtado-Ziola N, et al (2012) Production platforms for biotherapeutic glycoproteins. Occurrence, impact, and challenges of non-human sialylation. Biotechnol Genet Eng Rev 28:147–175
Dumont J, Euwart D, Mei B, et al (2015) Human cell lines for biopharmaceutical manufacturing: history, status, and future perspectives. Crit Rev Biotechnol:1–13. doi:10.3109/07388551.2015.1084266
Swiech K, Picanco-Castro V, Covas DT (2012) Human cells: new platform for recombinant therapeutic protein production. Protein Expression Purif 84(1):147–153. doi:10.1016/j.pep.2012.04.023
Havenga MJ, Holterman L, Melis I, et al (2008) Serum-free transient protein production system based on adenoviral vector and PER.C6 technology: high yield and preserved bioactivity. Biotechnol Bioeng 100(2):273–283. doi:10.1002/bit.21757
Schiedner G, Hertel S, Bialek C, et al (2008) Efficient and reproducible generation of high-expressing, stable human cell lines without need for antibiotic selection. BMC Biotechnol 8:13. doi:10.1186/1472-6750-8-13
Baumeister H, Goletz S (2010) A matter of cell line development. Eur Biopharm Rev 2010:54
Eisner F, Pichler M, Goletz S, et al (2015) A glyco-engineered anti-HER2 monoclonal antibody (TrasGEX) induces a long-lasting remission in a patient with HER2 overexpressing metastatic colorectal cancer after failure of all available treatment options. J Clin Pathol 68(12):1044–1046. doi:10.1136/jclinpath-2015-202996
Blanchard V, Liu X, Eigel D, Kaup M, Rieck S, Janciauskiene S, Sandig V, Marx U, Walden P, Tauber R, Berger M (2011) N-Glycosylation and biological activity of recombinant human alpha 1-antitrypsin expressed in a novel human neuronal cell line. Biotechnol Bioeng 108(9):2118–2128. doi:10.1002/bit.23158
Walsh G (2014) Biopharmaceutical benchmarks 2014. Nat Biotechnol 32(10):992–1000. doi:10.1038/nbt.3040
Berting A, Farcet MR, Kreil TR (2010) Virus susceptibility of Chinese hamster ovary (CHO) cells and detection of viral contaminations by adventitious agent testing. Biotechnol Bioeng 106(4):598–607. doi:10.1002/bit.22723
Kishishita S, Katayama S, Kodaira K, et al (2015) Optimization of chemically defined feed media for monoclonal antibody production in Chinese hamster ovary cells. J Biosci Bioeng 120(1):78–84. doi:10.1016/j.jbiosc.2014.11.022
Fischer S, Handrick R, Otte K (2015) The art of CHO cell engineering: a comprehensive retrospect and future perspectives. Biotechnol Adv 33(8):1878–1896. doi:10.1016/j.biotechadv.2015.10.015
Lewis NE, Liu X, Li Y, et al (2013) Genomic landscapes of Chinese hamster ovary cell lines as revealed by the Cricetulus griseus draft genome. Nat Biotechnol 31(8):759–765. doi:10.1038/nbt.2624
Xu X, Nagarajan H, Lewis NE, et al (2011) The genomic sequence of the Chinese hamster ovary (CHO)-K1 cell line. Nat Biotechnol 29(8):735–741. doi:10.1038/nbt.1932
Estes S, Melville M (2014) Mammalian cell line developments in speed and efficiency. Adv Biochem Eng Biotechnol 139:11–33. doi:10.1007/10_2013_260
Walther J, Godawat R, Hwang C, Abe Y, Sinclair A, Konstantinov K (2015) The business impact of an integrated continuous biomanufacturing platform for recombinant protein production. J Biotechnol 213:3–12. doi:10.1016/j.jbiotec.2015.05.010
Patil R, Walther J (2017) Continuous manufacturing of recombinant therapeutic proteins: upstream and downstream technologies. Adv Biochem Eng Biotechnol. doi:10.1007/10_2016_58
Wurm FM (2013) CHO quasispecies – implications for manufacturing processes. Processes 1:296–311
Tjio JH, Puck TT (1958) Genetics of somatic mammalian cells II. Chromosomal constitution of cells in tissue culture. J Exp Med 108(2):259–268
Hu Z, Guo D, Yip SS, et al (2013) Chinese hamster ovary K1 host cell enables stable cell line development for antibody molecules which are difficult to express in DUXB11-derived dihydrofolate reductase deficient host cell. Biotechnol Prog 29(4):980–985. doi:10.1002/btpr.1730
Kennard ML, Goosney DL, Monteith D, et al (2009) The generation of stable, high MAb expressing CHO cell lines based on the artificial chromosome expression (ACE) technology. Biotechnol Bioeng 104(3):540–553. doi:10.1002/bit.22406
Davies SL, Lovelady CS, Grainger RK, et al (2013) Functional heterogeneity and heritability in CHO cell populations. Biotechnol Bioeng 110(1):260–274. doi:10.1002/bit.24621
O’Callaghan PM, Berthelot ME, Young RJ, et al (2015) Diversity in host clone performance within a Chinese hamster ovary cell line. Biotechnol Prog 31(5):1187–1200. doi:10.1002/btpr.2097
Derouazi M, Martinet D, Besuchet Schmutz N, et al (2006) Genetic characterization of CHO production host DG44 and derivative recombinant cell lines. Biochem Biophys Res Commun 340(4):1069–1077
Patnaik SK, Stanley P (2006) Lectin-resistant CHO glycosylation mutants. Methods Enzymol 416:159–182
Bort JA, Stern B, Borth N (2010) CHO-K1 host cells adapted to growth in glutamine-free medium by FACS-assisted evolution. Biotechnol J 5(10):1090–1097. doi:10.1002/biot.201000095
Prentice HL, Ehrenfels BN, Sisk WP (2007) Improving performance of mammalian cells in fed-batch processes through “bioreactor evolution”. Biotechnol Prog 23(2):458–464. doi:10.1021/bp060296y
Jostock T, Knopf HP (2012) Mammalian stable expression of biotherapeutics. Methods Mol Biol 899:227–238. doi:10.1007/978-1-61779-921-1_15
Rita Costa A, Elisa Rodrigues M, Henriques M, et al (2010) Guidelines to cell engineering for monoclonal antibody production. Eur J Pharm Biopharm 74(2):127–138. doi:10.1016/j.ejpb.2009.10.002
Chusainow J, Yang YS, Yeo JH, Toh PC, Asvadi P, Wong NS, Yap MG (2009) A study of monoclonal antibody-producing CHO cell lines: what makes a stable high producer? Biotechnol Bioeng 102(4):1182–1196. doi:10.1002/bit.22158
Westwood AD, Rowe DA, Clarke HR (2010) Improved recombinant protein yield using a codon deoptimized DHFR selectable marker in a CHEF1 expression plasmid. Biotechnol Prog 26(6):1558–1566. doi:10.1002/btpr.491
Ng SK, Wang DI, Yap MG (2007) Application of destabilizing sequences on selection marker for improved recombinant protein productivity in CHO-DG44. Metab Eng 9(3):304–316
Ng SK, Tan TR, Wang Y, et al (2012) Production of functional soluble Dectin-1 glycoprotein using an IRES-linked destabilized-dihydrofolate reductase expression vector. PLoS One 7(12):e52785. doi:10.1371/journal.pone.0052785
Chin CL, Chin HK, Chin CS, et al (2015) Engineering selection stringency on expression vector for the production of recombinant human alpha1-antitrypsin using Chinese Hamster ovary cells. BMC Biotechnol 15:44. doi:10.1186/s12896-015-0145-9
Fan L, Kadura I, Krebs LE, et al (2012) Improving the efficiency of CHO cell line generation using glutamine synthetase gene knock-out cells. Biotechnol Bioeng 109(4):1007–1015. doi:10.1002/bit.24365
Roy G, Bowen MA (2015) Modulation of endogenous Glutamine Synthetase (GS) in CHO cells improves selection and characteristics of expression hosts. 12th Protein expression in animal cells conference
Curtin JA, Dane AP, Swanson A, et al (2008) Bidirectional promoter interference between two widely used internal heterologous promoters in a late-generation lentiviral construct. Gene Ther 15(5):384–390
Le H, Vishwanathan N, Kantardjieff A, et al (2013) Dynamic gene expression for metabolic engineering of mammalian cells in culture. Metab Eng 20:212–220. doi:10.1016/j.ymben.2013.09.004
Brown AJ, Sweeney B, Mainwaring DO, et al (2014) Synthetic promoters for CHO cell engineering. Biotechnol Bioeng 111(8):1638–1647. doi:10.1002/bit.25227
Ho SC, Bardor M, Li B, et al (2013) Comparison of internal ribosome entry site (IRES) and Furin-2A (F2A) for monoclonal antibody expression level and quality in CHO cells. PLoS One 8(5):e63247. doi:10.1371/journal.pone.0063247
Ho SC, Koh EY, van Beers M, et al (2013) Control of IgG LC:HC ratio in stably transfected CHO cells and study of the impact on expression, aggregation, glycosylation and conformational stability. J Biotechnol 165(3–4):157–166. doi:10.1016/j.jbiotec.2013.03.019
Ho SC, Mariati YJH, et al (2015) Impact of using different promoters and matrix attachment regions on recombinant protein expression level and stability in stably transfected CHO cells. Mol Biotechnol 57(2):138–144. doi:10.1007/s12033-014-9809-2
Ho SC, Wang T, Song Z, et al (2015) IgG aggregation mechanism for CHO cell lines expressing excess heavy chains. Mol Biotechnol 57(7):625–634. doi:10.1007/s12033-015-9852-7
Koh EYC, Ho SCL, Mariati et al. (2013) An Internal Ribosome Entry Site (IRES) mutant mlibrary for tuning expression level of multiple genes in mammalian cells. PLoS One 8(12):e82100. doi:10.1371/journal.pone.0082100
Chng J, Wang T, Nian R, et al (2015) Cleavage efficient 2A peptides for high level monoclonal antibody expression in CHO cells. mAbs 7(2):403–412. doi:10.1080/19420862.2015.1008351
Airenne KJ, Hu YC, Kost TA, et al (2013) Baculovirus: an insect-derived vector for diverse gene transfer applications. Mol Ther 21(4):739–749. doi:10.1038/mt.2012.286
Mufarrege EF, Antuna S, Etcheverrigaray M, et al (2014) Development of lentiviral vectors for transient and stable protein overexpression in mammalian cells. A new strategy for recombinant human FVIII (rhFVIII) production. Protein Expression Purif 95:50–56. doi:10.1016/j.pep.2013.11.005
Harraghy N, Regamey A, Girod PA, et al (2011) Identification of a potent MAR element from the mouse genome and assessment of its activity in stable and transient transfections. J Biotechnol 154(1):11–20. doi:10.1016/j.jbiotec.2011.04.004
Antoniou M, Harland L, Mustoe T, et al (2003) Transgenes encompassing dual-promoter CpG islands from the human TBP and HNRPA2B1 loci are resistant to heterochromatin-mediated silencing. Genomics 82(3):269–279. doi:10.1016/s0888-7543(03)00107-1
Betts Z, Croxford AS, Dickson AJ (2015) Evaluating the interaction between UCOE and DHFR-linked amplification and stability of recombinant protein expression. Biotechnol Prog 31(4):1014–1025. doi:10.1002/btpr.2083
Dharshanan S, Chong H, Cheah SH, et al (2014) Stable expression of H1C2 monoclonal antibody in NS0 and CHO cells using pFUSE and UCOE expression system. Cytotechnology 66(4):625–633. doi:10.1007/s10616-013-9615-x
Hou JJ, Hughes BS, Smede M, et al (2014) High-throughput ClonePix FL analysis of mAb-expressing clones using the UCOE expression system. New Biotechnol 31(3):214–220. doi:10.1016/j.nbt.2014.02.002
Liu H, Wang X, Shi S, Chen Y, Han W (2015) Efficient production of FAM19A4, a novel potential cytokine, in a stable optimized CHO-S cell system. Protein Expr Purif 113:1–7. doi:10.1016/j.pcp.2015.05.004
Saunders F, Sweeney B, Antoniou MN, et al (2015) Chromatin function modifying elements in an industrial antibody production platform–comparison of UCOE, MAR, STAR and cHS4 elements. PLoS One 10(4):e0120096. doi:10.1371/journal.pone.0120096
Ivics Z, Li MA, Mates L, et al (2009) Transposon-mediated genome manipulation in vertebrates. Nat Methods 6(6):415–422. doi:10.1038/nmeth.1332
Alattia JR, Matasci M, Dimitrov M, et al (2013) Highly efficient production of the Alzheimer’s ϒ-Secretase integral membrane protease complex by a multi-gene stable integration approach. Biotechnol Bioeng 110(7):1995–2005. doi:10.1002/bit.24851
Li MA, Turner DJ, Ning Z, et al (2011) Mobilization of giant piggyBac transposons in the mouse genome. Nucleic Acids Res 39(22):e148. doi:10.1093/nar/gkr764
Matasci M, Baldi L, Hacker DL, et al (2011) The PiggyBac transposon enhances the frequency of CHO stable cell line generation and yields recombinant lines with superior productivity and stability. Biotechnol Bioeng 108(9):2141–2150. doi:10.1002/bit.23167
Balasubramanian S, Matasci M, Kadlecova Z, et al (2015) Rapid recombinant protein production from piggyBac transposon-mediated stable CHO cell pools. J Biotechnol 200:61–69. doi:10.1016/j.jbiotec.2015.03.001
Ley D, Harraghy N, Le Fourn V, et al (2013) MAR elements and transposons for improved transgene integration and expression. PLoS One 8(4):e62784. doi:10.1371/journal.pone.0062784
Nehlsen K, Schucht R, da Gama-Norton L, et al (2009) Recombinant protein expression by targeting pre-selected chromosomal loci. BMC Biotechnol 9:100. doi:10.1186/1472-6750-9-100
Bode J, Schlake T, Iber M, et al (2000) The transgeneticist’s toolbox: novel methods for the targeted modification of eukaryotic genomes. Biol Chem 381(9–10):801–813. doi:10.1515/BC.2000.103
Turan S, Zehe C, Kuehle J, et al (2013) Recombinase-mediated cassette exchange (RMCE) – a rapidly-expanding toolbox for targeted genomic modifications. Gene 515(1):1–27. doi:10.1016/j.gene.2012.11.016
Kim MS, Lee GM (2008) Use of Flp-mediated cassette exchange in the development of a CHO cell line stably producing erythropoietin. J Microbiol Biotechnol 18(7):1342–1351
Kito M, Itami S, Fukano Y, et al (2002) Construction of engineered CHO strains for high-level production of recombinant proteins. Appl Microbiol Biotechnol 60(4):442–448. doi:10.1007/s00253-002-1134-1
Crawford Y, Zhou M, Hu Z, et al (2013) Fast identification of reliable hosts for targeted cell line development from a limited-genome screening using combined ϕC31 integrase and CRE-Lox technologies. Biotechnol Prog 29(5):1307–1315. doi:10.1002/btpr.1783
Zhang L, Inniss MC, Han S, et al (2015) Recombinase-mediated cassette exchange (RMCE) for monoclonal antibody expression in the commercially relevant CHOK1SV cell line. Biotechnol Prog 31(6):1645–1656. doi:10.1002/btpr.2175
Chandrasegaran S, Carroll D (2015) Origins of programmable nucleases for genome engineering. J Mol Biol 428:963. doi:10.1016/j.jmb.2015.10.014
Cristea S, Freyvert Y, Santiago Y, et al (2013) In vivo cleavage of transgene donors promotes nuclease-mediated targeted integration. Biotechnol Bioeng 110(3):871–880. doi:10.1002/bit.24733
Sakuma T, Nakade S, Sakane Y, et al (2016) MMEJ-assisted gene knock-in using TALENs and CRISPR-Cas9 with the PITCh systems. Nat Protoc 11(1):118–133. doi:10.1038/nprot.2015.140
Sakuma T, Takenaga M, Kawabe Y, et al (2015) Homologous recombination-independent large gene cassette knock-in in CHO cells using TALEN and MMEJ-directed donor plasmids. Int J Mol Sci 16(10):23849–23866. doi:10.3390/ijms161023849
Lee JS, Grav LM, Lewis NE, et al (2015) CRISPR/Cas9-mediated genome engineering of CHO cell factories: application and perspectives. Biotechnol J 10(7):979–994. doi:10.1002/biot.201500082
Lee JS, Kallehauge TB, Pedersen LE, et al (2015) Site-specific integration in CHO cells mediated by CRISPR/Cas9 and homology-directed DNA repair pathway. Sci Rep 5:8572. doi:10.1038/srep08572
Bachu R, Bergareche I, Chasin LA (2015) CRISPR-Cas targeted plasmid integration into mammalian cells via non-homologous end joining. Biotechnol Bioeng 112(10):2154–2162. doi:10.1002/bit.25629
Hossler P, Khattak SF, Li ZJ (2009) Optimal and consistent protein glycosylation in mammalian cell culture. Glycobiology 19(9):936–949. doi:10.1093/glycob/cwp079
Jiang XR, Song A, Bergelson S, Arroll T, Parekh B, May K, Chung S, Strouse R, Mire-Sluis A, Schenerman M (2011) Advances in the assessment and control of the effector functions of therapeutic antibodies. Nat Rev Drug Discovery 10(2):101–111. doi:10.1038/nrd3365
Dicker M, Strasser R (2015) Using glyco-engineering to produce therapeutic proteins. Expert Opin Biol Ther 15(10):1501–1516. doi:10.1517/14712598.2015.1069271
Meuris L, Santens F, Elson G, et al (2014) GlycoDelete engineering of mammalian cells simplifies N-glycosylation of recombinant proteins. Nat Biotechnol 32(5):485–489. doi:10.1038/nbt.2885
Monaco L, Marc A, Eon-Duval A, et al (1996) Genetic engineering of α2,6-sialyltransferase in recombinant CHO cells and its effects on the sialylation of recombinant interferon-γ. Cytotechnology 22(1–3):197–203. doi:10.1007/BF00353939
Weikert S, Papac D, Briggs J, et al (1999) Engineering Chinese hamster ovary cells to maximize sialic acid content of recombinant glycoproteins. Nat Biotechnol 17(11):1116–1121
Lin N, Mascarenhas J, Sealover NR, et al (2015) Chinese hamster ovary (CHO) host cell engineering to increase sialylation of recombinant therapeutic proteins by modulating sialyltransferase expression. Biotechnol Prog 31(2):334–346. doi:10.1002/btpr.2038
Yin B, Gao Y, Chung CY, Blake E, Stuczynski MC, Tang J, Kildegaard HF, Andersen MR, Zhang H, Betenbaugh MJ (2015) Glycoengineering of Chinese hamster ovary cells for enhanced erythropoietin N-glycan branching and sialylation. Biotechnol Bioeng 112(11):2343–2351. doi:10.1002/bit.25650
Chung CY, Yin B, Wang Q, Chuang KY, Chu JH, Betenbaugh MJ (2015) Assessment of the coordinated role of ST3GAL3, ST3GAL4 and ST3GAL6 on the α2,3 sialylation linkage of mammalian glycoproteins. Biochem Biophys Res Common 463(3):211–215
Goetze AM, Liu YD, Zhang Z, et al (2011) High-mannose glycans on the Fc region of therapeutic IgG antibodies increase serum clearance in humans. Glycobiology 21(7):949–959. doi:10.1093/glycob/cwr027
Oganesyan V, Mazor Y, Yang C, et al (2015) Structural insights into the interaction of human IgG1 with FcgammaRI: no direct role of glycans in binding. Acta Crystallogr Sect D Biol Crystallogr 71(Pt 11):2354–2361. doi:10.1107/S1399004715018015
Wilke S, Groebe L, Maffenbeier V, et al (2011) Streamlining homogeneous glycoprotein production for biophysical and structural applications by targeted cell line development. PLoS One 6(12):e27829. doi:10.1371/journal.pone.0027829
Zhong X, Cooley C, Seth N, et al (2012) Engineering novel Lec1 glycosylation mutants in CHO-DUKX cells: molecular insights and effector modulation of N-acetylglucosaminyltransferase I. Biotechnol Bioeng 109(7):1723–1734. doi:10.1002/bit.24448
Sealover NR, Davis AM, Brooks JK, et al (2013) Engineering Chinese Hamster Ovary (CHO) cells for producing recombinant proteins with simple glycoforms by zinc-finger nuclease (ZFN) – mediated gene knock-out of mannosyl (alpha-1,3-)-glycoprotein beta-1,2-N-acetylglucosaminyltransferase (Mgat1). J Biotechnol 167(1):24–32. doi:10.1016/j.jbiotec.2013.06.006
Van Patten SM, Hughes H, Huff MR, et al (2007) Effect of mannose chain length on targeting of glucocerebrosidase for enzyme replacement therapy of Gaucher disease. Glycobiology 17(5):467–478. doi:10.1093/glycob/cwm008
Goh JS, Liu Y, Chan KF, et al (2014) Producing recombinant therapeutic glycoproteins with enhanced sialylation using CHO-gmt4 glycosylation mutant cells. Bioengineered 5(4):269–273. doi:10.4161/bioe.29490
Goh JS, Liu Y, Liu H, et al (2014) Highly sialylated recombinant human erythropoietin production in large-scale perfusion bioreactor utilizing CHO-gmt4 (JW152) with restored GnT I function. Biotechnol J 9(1):100–109. doi:10.1002/biot.201300301
Lin N, Davis D, Sealover NR et al (2013) Mgat4 may play a role in increased sialylation by overexpressing functional MGAT1 in Mgat1-disrupted Chinese Hamster Ovary (CHO) cells. In: Anonymous bioprocess international conference and exhibition 2013, Boston, MA, Sept 2013
Yamane-Ohnuki N, Kinoshita S, Inoue-Urakubo M, et al (2004) Establishment of FUT8 knock-out Chinese hamster ovary cells: an ideal host cell line for producing completely defucosylated antibodies with enhanced antibody-dependent cellular cytotoxicity. Biotechnol Bioeng 87(5):614–622. doi:10.1002/bit.20151
Malphettes L, Freyvert Y, Chang J, et al (2010) Highly efficient deletion of FUT8 in CHO cell lines using zinc-finger nucleases yields cells that produce completely nonfucosylated antibodies. Biotechnol Bioeng 106(5):774–783. doi:10.1002/bit.22751
Grav LM, Lee JS, Gerling S, et al (2015) One-step generation of triple knock-out CHO cell lines using CRISPR/Cas9 and fluorescent enrichment. Biotechnol J 10(9):1446–1456. doi:10.1002/biot.201500027
Haryadi R, Zhang P, Chan KF, et al (2013) CHO-gmt5, a novel CHO glycosylation mutant for producing afucosylated and asialylated recombinant antibodies. Bioengineered 4(2):90–94. doi:10.4161/bioe.22262
von Horsten HH, Ogorek C, Blanchard V, et al (2010) Production of non-fucosylated antibodies by co-expression of heterologous GDP-6-deoxy-D-lyxo-4-hexulose reductase. Glycobiology 20(12):1607–1618. doi:10.1093/glycob/cwq109
Yang Z, Halim A, Narimatsu Y, et al (2014) The GalNAc-type O-glycoproteome of CHO cells characterized by the SimpleCell strategy. Mol Cell Proteomics 13(12):3224–3235. doi:10.1074/mcp.M114.041541
Estes B, Hsu YR, Tam LT, et al (2015) Uncovering methods for the prevention of protein aggregation and improvement of product quality in a transient expression system. Biotechnol Prog 31(1):258–267. doi:10.1002/btpr.2021
Le Fourn V, Girod PA, Buceta M, et al (2014) CHO cell engineering to prevent polypeptide aggregation and improve therapeutic protein secretion. Metab Eng 21:91–102. doi:10.1016/j.ymben.2012.12.003
Pybus LP, Dean G, West NR, et al (2014) Model-directed engineering of “difficult-to-express” monoclonal antibody production by Chinese hamster ovary cells. Biotechnol Bioeng 111(2):372–385. doi:10.1002/bit.25116
Johari YB, Estes SD, Alves CS, et al (2015) Integrated cell and process engineering for improved transient production of a “difficult-to-express” fusion protein by CHO cells. Biotechnol Bioeng 112(12):2527–2542. doi:10.1002/bit.25687
Gulis G, Simi KC, de Toledo RR, Maranhao Q, Brigido MM (2014) Optimization of heterologous protein production in Chinese hamster ovary cues under overexpression of spliced form of human X-box binding protein. BMC Biotechnol 14:26. doi:10.1186/1472-6750-14-26
Misaghi S, Chang J, Snedecor B (2014) It’s time to regulate: coping with product-induced nongenetic clonal instability in CHO cell lines via regulated protein expression. Biotechnol Prog 30(6):1432–1440. doi:10.1002/btpr.1970
Romand S, Jostock T, Fornaro M, et al (2015) Improving expression of recombinant human IGF-1 using IGF-1R knock-out CHO cell lines. Biotechnol Bioeng 113:1094. doi:10.1002/bit.25877
Alves CS, Gilbert A, Dalvi S, et al (2015) Integration of cell line and process development to overcome the challenge of a difficult to express protein. Biotechnol Prog 31(5):1201–1211. doi:10.1002/btpr.2091
Chakrabarti A, Chen AW, Varner JD (2011) A review of the mammalian unfolded protein response. Biotechnol Bioeng 108(12):2777–2793. doi:10.1002/bit.23282
Du Z, Treiber D, McCoy RE, et al (2013) Non-invasive UPR monitoring system and its applications in CHO production cultures. Biotechnol Bioeng 110(8):2184–2194. doi:10.1002/bit.24877
Kober L, Zehe C, Bode J (2012) Development of a novel ER stress based selection system for the isolation of highly productive clones. Biotechnol Bioeng 109(10):2599–2611. doi:10.1002/bit.24527
Kober L, Zehe C, Bode J (2013) Optimized signal peptides for the development of high expressing CHO cell lines. Biotechnol Bioeng 110(4):1164–1173. doi:10.1002/bit.24776
Ishii Y, Murakami J, Sasaki K, et al (2014) Efficient folding/assembly in Chinese hamster ovary cells is critical for high quality (low aggregate content) of secreted trastuzumab as well as for high production: stepwise multivariate regression analyses. J Biosci Bioeng 118(2):223–230. doi:10.1016/j.jbiosc.2014.01.013
Onitsuka M, Kawaguchi A, Asano R, et al (2014) Glycosylation analysis of an aggregated antibody produced by Chinese hamster ovary cells in bioreactor culture. J Biosci Bioeng 117(5):639–644. doi:10.1016/j.jbiosc.2013.11.001
Gomez N, Subramanian J, Ouyang J, et al (2012) Culture temperature modulates aggregation of recombinant antibody in cho cells. Biotechnol Bioeng 109(1):125–136. doi:10.1002/bit.23288
Han YK, Koo TY, Lee GM (2009) Enhanced interferon-beta production by CHO cells through elevated osmolality and reduced culture temperature. Biotechnol Prog 25(5):1440–1447. doi:10.1002/btpr.234
Hansen HG, Nilsson CN, Lund AM, Kol S, Grav LM, Lundqvist M, Roclberg J, Lee GM, Andersen MR, Kildegaard HF (2015) Versatile microscale screening platform for improving recombinant protein productivity in Chinese hamster ovary cells. Sci Rep 5:18016. doi:10.1038/srep18016
Geisse S, Voedisch B (2012) Transient expression technologies: past, present, and future. Methods Mol Biol 899:203–219. doi:10.1007/978-1-61779-921-1_13
Bohm E, Seyfried BK, Dockal M, et al (2015) Differences in N-glycosylation of recombinant human coagulation factor VII derived from BHK, CHO, and HEK293 cells. BMC Biotechnol 15:87. doi:10.1186/s12896-015-0205-1
Croset A, Delafosse L, Gaudry JP, et al (2012) Differences in the glycosylation of recombinant proteins expressed in HEK and CHO cells. J Biotechnol 161(3):336–348. doi:10.1016/j.jbiotec.2012.06.038
Rajendra Y, Balasubramanian S, Kiseljak D, Baldi L, Wurm FM, Hacker DL (2015) Enhanced plasmid DNA utilization in transiently transfected CHO-DG44 cells in the presence of polar solvents. Biotechnol Prog 31(6):1571–1578. doi:10.1002/btpr.2152
Rajendra Y, Hougland MD, Alam R, et al (2015) A high cell density transient transfection system for therapeutic protein expression based on a CHO GS-knock-out cell line: process development and product quality assessment. Biotechnol Bioeng 112(5):977–986. doi:10.1002/bit.25514
Jager V, Bussow K, Schirrmann T (2015) Transient recombinant protein expression in mammalian cells. In: Al-Rubeai M (ed) Animal cell culture, Cell engineering, vol 9. Springer, Cham. doi:10.1007/978-3-319-10320-4_2
Codamo J, Munro TP, Hughes BS, Song M, Gray PP (2011) Enhanced CHO cell-based transient gene expression with the epi-CHO expression system. Mol Biotechnol 48(2):109–115. doi:10.1007/s12033-010-9351-9
Kunaparaju R, Liao M, Sunstrom NA (2005) Epi-CHO, an episomal expression system for recombinant protein production in CHO cells. Biotechnol Bioeng 91(6):670–67677
Daramola O, Stevenson J, Dean G, et al (2014) A high-yielding CHO transient system: coexpression of genes encoding EBNA-1 and GS enhances transient protein expression. Biotechnol Prog 30(1):132–141. doi:10.1002/btpr.1809
Cain K, Peters S, Hailu H, Sweeney B, Stephens P, Heads J, Sarkar K, Ventom A, Page C, Dickson A (2013) A CHO cell line engineered to express XBP1 and ERO-Lα has increased levels of transient protein expression. Biotechnol Prog 29(3):697–706. doi:10.1002/btpr.1693
Rajendra Y, Kiseljak D, Baldi L, Hacker DL, Wurm FM (2011) A simple high-yielding process for transient gene expression in CHO cells. J Biotechnol 153(1–2):22–26. doi:10.1016/j.jbiotec.2011.03.001
Rajendra Y, Kiseljak D, Baldi L, Hacker DL, Wurm FM (2012) Reduced glutamine concentration improves protein production in growth-arrested CHO-DG44 and HEK-293E cells. Biotechnol Lett 34(4):619–626. doi:10.1007/s10529-011-0809-z
International Committee for Harmonization Topic Q5D (ICH Q5D) (1997) Derivation and characterisation of cell substrates used for production of biotechnological/biological products (CPMP/ICH/294/95). International conference on harmonisation of technical requirements for registration of pharmaceuticals for human use
Nakamura T, Omasa T (2015) Optimization of cell line development in the GS-CHO expression system using a high-throughput, single cell-based clone selection system. J Biosci Bioeng 120(3):323–329. doi:10.1016/j.jbiosc.2015.01.002
Evans K, Albanetti T, Venkat R, et al (2015) Assurance of monoclonality in one round of cloning through cell sorting for single cell deposition coupled with high resolution cell imaging. Biotechnol Prog 31(5):1172–1178. doi:10.1002/btpr.2145
Mazutis L, Gilbert J, Ung WL, et al (2013) Single-cell analysis and sorting using droplet-based microfluidics. Nat Protoc 8(5):870–891. doi:10.1038/nprot.2013.046
Joensson HN, Zhang C, Uhlen M, et al (2012) A homogeneous assay for protein analysis in droplets by fluorescence polarization. Electrophoresis 33(3):436–439. doi:10.1002/elps.201100350
Gross A, Schondube J, Niekrawitz S, et al (2013) Single-cell printer: automated, on demand, and label free. J Lab Autom 18(6):504–518. doi:10.1177/2211068213497204
Stumpf F, Schoendube J, Gross A, et al (2015) Single-cell PCR of genomic DNA enabled by automated single-cell printing for cell isolation. Biosens Bioelectron 69:301–306. doi:10.1016/j.bios.2015.03.008
Silk NJ, Denby S, Lewis G, et al (2010) Fed-batch operation of an industrial cell culture process in shaken microwells. Biotechnol Lett 32(1):73–78. doi:10.1007/s10529-009-0124-0
Hsu WT, Aulakh RP, Traul DL, et al (2012) Advanced microscale bioreactor system: a representative scale-down model for bench-top bioreactors. Cytotechnology 64(6):667–678. doi:10.1007/s10616-012-9446-1
Rameez S, Mostafa SS, Miller C, et al (2014) High-throughput miniaturized bioreactors for cell culture process development: reproducibility, scalability, and control. Biotechnol Prog 30(3):718–727. doi:10.1002/btpr.1874
Paul AJ, Schwab K, Hesse F (2014) Direct analysis of mAb aggregates in mammalian cell culture supernatant. BMC Biotechnol 14:99. doi:10.1186/s12896-014-0099-3
Reusch D, Haberger M, Selman MH, et al (2013) High-throughput work flow for IgG Fc-glycosylation analysis of biotechnological samples. Anal Biochem 432(2):82–89. doi:10.1016/j.ab.2012.09.032
Yang Y, Strahan A, Li C, et al (2010) Detecting low level sequence variants in recombinant monoclonal antibodies. mAbs 2(3):285–298. doi:10.4161/mabs.2.3.11718
Ambrogelly A, Liu YH, Li H, et al (2012) Characterization of antibody variants during process development: the tale of incomplete processing of N-terminal secretion peptide. mAbs 4(6):701–709. doi:10.4161/mabs.21614
Harris RP, Kilby PM (2014) Amino acid misincorporation in recombinant biopharmaceutical products. Curr Opin Biotechnol 30:45–50. doi:10.1016/j.copbio.2014.05.003
Khetan A, Huang YM, Dolnikova J, et al (2010) Control of misincorporation of serine for asparagine during antibody production using CHO cells. Biotechnol Bioeng 107(1):116–123. doi:10.1002/bit.22771
Kaas CS, Kristensen C, Betenbaugh MJ, et al (2015) Sequencing the CHO DXB11 genome reveals regional variations in genomic stability and haploidy. BMC Genomics 16:160. doi:10.1186/s12864-015-1391-x
Wright C, Groot J, Swahn S, McLaughlin H, Liu M, Xu C, Sun C, Zheng E, Estes S (2016) Genetic mutation analysis at early stages of cell line development using next generation sequencing. Biotechnol Prog 32(3):813–817. doi:10.1002/btpr.2263
Zhang S, Bartkowiak L, Nabiswa B, Mishra P, Fann J, Ouelette D, Correia I, Da R, Liu J (2015) Identifying low-level sequence variants via next generation sequencing to aid stable CHO cell line screening. Biotechnol Prog 31(4):1077–1085
Kremkow BG, Lee KH (2015) Sequencing technologies for animal cell culture research. Biotechnol Lett 37(1):55–65. doi:10.1007/s10529-014-1660-9
de Vree PJ, de Wit E, Yilmaz M, et al (2014) Targeted sequencing by proximity ligation for comprehensive variant detection and local haplotyping. Nat Biotechnol 32(10):1019–1025. doi:10.1038/nbt.2959
Cabannes E, Hebert C, Eloit M (2014) Whole genome: next-generation sequencing as a virus safety test for biotechnological products. PDA J Pharm Sci Technol 68(6):631–638. doi:10.5731/pdajpst.2014.01015
Khan AS, Vacante DA (2014) Advanced technologies for virus detection in the evaluation of biologicals- applications and challenges. PDA Journal Pharm Sci and Technol 68:546–547. doi:10.5731/pdajpst.2014.01028
Mee ET, Preston MD, Minor PD, Schepelmann S (2016) Development of a candidate reference material for adventitious virus detection in vaccine and biologicals manufacturing by deep sequencing. Vaccine 12(34):2035. doi:10.1016/j.vaccine.2015.12.020
Laux H, Romand S, Ritter A, Oertli M, Fornaro M, Jostock T, Wilms B (2013) Generation of genetically engineered CHO cell lines to support the production of a difficult to express therapeutic protein. BMC Proc 7:P1
Kildegaard HF, Baycin-Hizal D, Lewis NE, et al (2013) The emerging CHO systems biology era: harnessing the ‘omics revolution for biotechnology. Curr Opin Biotechnol 24(6):1102–1107. doi:10.1016/j.copbio.2013.02.007
Gutierrez JM, Lewis NE (2015) Optimizing eukaryotic cell hosts for protein production through systems biotechnology and genome-scale modeling. Biotechnol J 10(7):939–949. doi:10.1002/biot.201400647
Jadhav V, Hackl M, Druz A, et al (2013) CHO microRNA engineering is growing up: recent successes and future challenges. Biotechnol Adv 31(8):1501–1513. doi:10.1016/j.biotechadv.2013.07.007
Fischer S, Buck T, Wagner A, et al (2014) A functional high-content miRNA screen identifies miR-30 family to boost recombinant protein production in CHO cells. Biotechnol J 9(10):1279–1292. doi:10.1002/biot.201400306
Jadhav V, Hackl M, Klanert G, et al (2014) Stable overexpression of miR-17 enhances recombinant protein production of CHO cells. J Biotechnol 175:38–44. doi:10.1016/j.jbiotec.2014.01.032
Lienert F, Lohmueller JJ, Abhishek G, Silver PA (2014) Synthetic biology in mammalian cells: next generation research tools and therapeutics. Nat Rev Mol Cell Biol 15:95–107. doi:10.1038/nrm3738
Xie M, Fussenegger M (2015) Mammalian designer cells: engineering principles and biomedical applications. Biotechnol J 10(7):1005–1018. doi:10.1002/biot.201400642
Brown AJ, James DC (2016) Precision control of recombinant gene transcription for CHO cell synthetic biology. Biotechnol Adv 34(5):492–503. doi:10.1016/jbiotechadv.2015.12.012
Lund AM, Kildegaard HF, Petersen MB, et al (2014) A versatile system for USER cloning-based assembly of expression vectors for mammalian cell engineering. PLoS One 9(5):e96693. doi:10.1371/journal.pone.0096693
Nielsen L, Borth N (2015) Editorial: on the cusp of rational CHO cell engineering. Biotechnol J 10(7):929–930. doi:10.1002/biot.201500375
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Zhu, J., Hatton, D. (2017). New Mammalian Expression Systems. In: Kiss, B., Gottschalk, U., Pohlscheidt, M. (eds) New Bioprocessing Strategies: Development and Manufacturing of Recombinant Antibodies and Proteins. Advances in Biochemical Engineering/Biotechnology, vol 165. Springer, Cham. https://doi.org/10.1007/10_2016_55
Download citation
DOI: https://doi.org/10.1007/10_2016_55
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-97108-7
Online ISBN: 978-3-319-97110-0
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)